In the process of primary cell culture, the cells acquired were round with bright and clear boundaries before the attached state. As the neurons grew, dendrites and axons started to be distinct. After 5–7 days of culture, the neurons reached a mature form with long projections, which were ideal for imaging or function studies. Although most of the impurities and cell debris could be removed due to changing media, certain residuals attached to poly-D-lysine and laminin coating were visible (Figure 1).
After proper culture, neurons could be identified via typical β-III-tubulin and MAP-2 immunostaining10,12. In addition, glia was specifically identified via GFAP immunostaining10. Mature neurons developed synaptic spines, which were close to the presynaptic specializations identified by the immunostaining of the synaptic protein maker, synapsin-1 (Figure 2)12. These results indicated that mature cells with well-developed synapses were obtained through this method. This result suggests its important role in future function studies.
Meanwhile, several neuron subtypes were recognized through immunocytochemistry experiments (Figure 3). Peptidergic neurons, which contain various neuropeptides, were immunostained with substance P13. Purinergic neurons with expressed vesicular nucleotide transporters were identified via SLC17A9 staining14. Nitrergic neurons were visualized with DYNLL-2, which connects nNOS with motor proteins in neurons15. Cholinergic neurons were immunoreactive with choline acetyltransferase16.
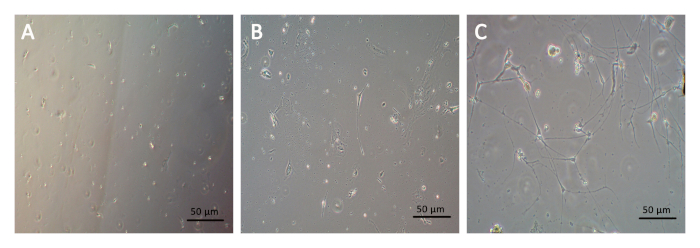
Figure 1. Phase-contrast images of primary cells isolated from the rat bladder culture taken at 1, 3, and 7 days after plating (A, B, C, respectively). Scale bar: 50 μm. Please click here to view a larger version of this figure.
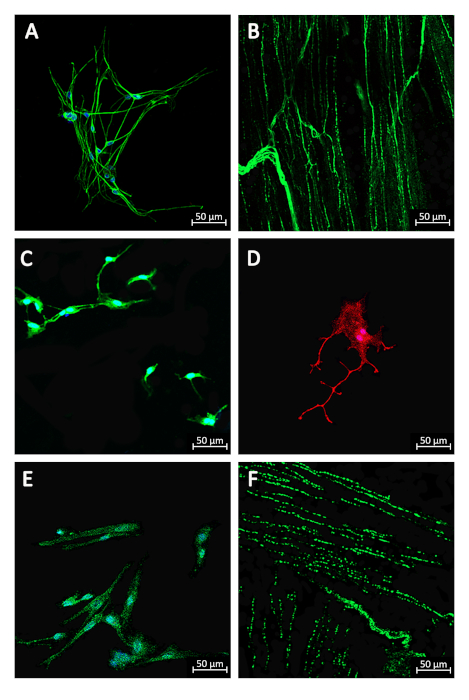
Figure 2. Immunofluorescence images of primary cells isolated from the rat bladder. Confocal microscopy analysis showed neuron cytoskeleton protein staining (β-III-tubulin, RRID: AB_2827688, 1:200) in primary culture neurons (A) and in whole mount bladder preparation (B). In primary culture neurons, neuronal phosphoprotein immunostaining (MAP 2, RRID:AB_2827689, 1:200) was also visualized (C). Glia were identified via glial fibrillary acidic protein staining (D; RRID: AB_627673, 1:50). Synapsins were visualized via synapsin protein staining (Synapsin-1, RRID: AB_2798146, 1:200) in cellular (E) and tissue (F) levels. The secondary antibodies used were as follows: Alexa Fluor 488 (green, goat anti-rabbit lgG, 1:200), Alexa Fluor 555 (red, goat anti-mouse lgG, 1:200). The nucleus was visualized using Hoechst 33342 (A, C, D, E; blue, 1 μg/mL). Scale bar: 50 μm. Please click here to view a larger version of this figure.
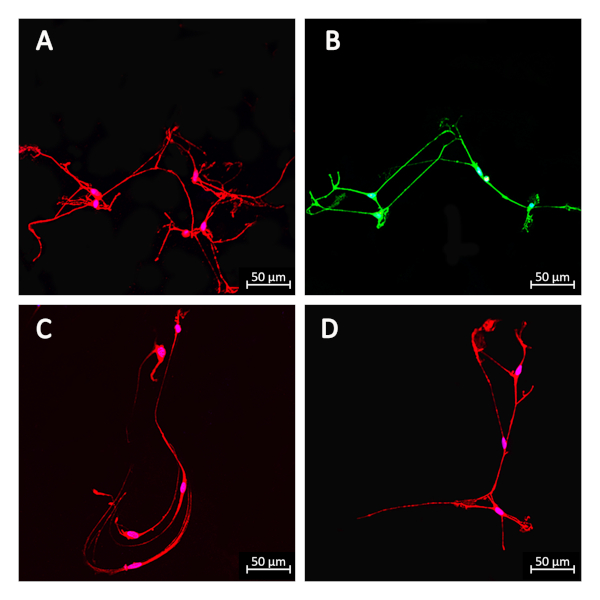
Figure 3. Immunofluorescence images of several neuron subtypes of primary neurons. Peptidergic neurons were immunostained with substance P (A; RRID: AB_785913, 1:50). Purinergic neurons were identified via SLC17A9 staining (B; RRID: AB_10597575, 1:200). Nitrergic neurons were visualized via DYNLL-2 staining (C; RRID: AB_654147, 1:50). Cholinergic neurons were immunoreactive with choline acetyltransferase (D; RRID: AB_2244867, 1:100). The secondary antibodies used were as follows: Alexa Fluor 488 (green, goat anti-rabbit lgG, 1:200), Alexa Fluor 555 (red, goat anti-mouse lgG, 1:200). The nucleus was visualized using Hoechst 33342 (A, B, C, D, blue, 1 μg/mL). Scale bar: 50 μm. Please click here to view a larger version of this figure.
| Ingredients | Molarity (mM) |
| NaCI | 120 |
| KCI | 5.9 |
| NaHCO3 | 25 |
| Na2HPO4·12H2O | 1.2 |
| MgCI2·6H2O | 1.2 |
| CaCI2 | 2.5 |
| Glucose | 11.5 |
Table 1. Krebs solution composition
