Vätskor och smälter är aktiva kemiska och fysiska transportvektorer i naturliga miljöer. De förhöjda frekvenserna av atomisk diffusion gynnar kemiska utbyten och reaktioner, den låga viskositeten i kombination med varierande flytkraft gynnar stor massöverföring, och kristallsmälttäthetsrelationer gynnar skiktning inuti planetariska kroppar. Frånvaron av ett periodiskt gitter, typiska höga temperaturer som krävs för att nå det smälta tillståndet och svårigheten för släckning gör den experimentella bestämningen av en serie uppenbara egenskaper, som densitet, diffusion och viskositet, extremt utmanande. Dessa svårigheter gör alternativa beräkningsmetoder starka och användbara verktyg för att undersöka denna klass av material.
Med tillkomsten av datorkraft och tillgången till superdatorer används för närvarande två stora numeriska atomistiska simuleringstekniker för att studera det dynamiska tillståndet hos ett icke-kristallint atomistiskt system, Monte Carlo1 och molekylär dynamik (MD)1,2. I Monte Carlo-simuleringar provtas det konfigurationsutrymmet slumpmässigt. Monte Carlo-metoder visar linjär skalning i parallellisering om alla provtagningsobservationer är oberoende av varandra. Resultatens kvalitet beror på kvaliteten på slumptalsgeneratorn och provtagningens representativitet. Monte Carlo-metoder visar linjär skalning i parallellisering om provtagningen är oberoende av varandra. I molekylär dynamik (MD) provtas det konfigurationsmässiga utrymmet av tidsberoende atombanor. Från en given konfiguration beräknas atombanorna genom att integrera de newtonska rörelseekvationerna. De interatomära krafterna kan beräknas med hjälp av modellinteratomiska potentialer (i klassisk MD) eller med hjälp av första principer metoder (i ab initio, eller första principer, MD). Kvaliteten på resultaten beror på banans längd och dess förmåga att inte lockas till lokala minima.
Molekyldynamiksimuleringar innehåller en mängd information, allt relaterat till systemets dynamiska beteende. Termodynamiska genomsnittliga egenskaper, som intern energi, temperatur och tryck, är ganska standard att beräkna. De kan extraheras från utdatafilerna för simuleringarna och medelvärdet, medan kvantiteter som är direkt relaterade till atomernas rörelse samt deras ömsesidiga förhållande måste beräknas efter extraktion av atompositioner och hastigheter.
Följaktligen har mycket arbete ägnats åt att visualisera resultaten, och olika paket finns tillgängliga idag, på olika plattformar, öppen källkod eller inte [Ovito3, VMD4, Vesta5, Travis6, etc.]. Alla dessa visualiseringsverktyg hanterar effektivt interatomära avstånd, och som sådan möjliggör de effektiv beräkning av parfördelningsfunktioner och diffusionskoefficienter. Olika grupper som utför storskaliga molekylära dynamiksimuleringar har proprietär programvara för att analysera olika andra egenskaper som härrör från simuleringarna, ibland i shareware eller andra former av begränsad tillgång till samhället, och ibland begränsad i omfattning och användning till vissa specifika paket. Sofistikerade algoritmer för att extrahera information om interatomär bindning, geometriska mönster och termodynamik utvecklas och implementeras i några av dessa paket3,4,5,6,7, etc.
Här föreslår vi UMD-paketet – ett paket med öppen källkod skrivet i Python för att analysera utdata från molekylära dynamiksimuleringar. UMD-paketet möjliggör beräkning av ett brett spektrum av strukturella, dynamiska och termodynamiska egenskaper (figur 1). Paketet är tillgängligt via GitHubs webbplats (https://github.com/rcaracas/UMD_package) och via en dedikerad sida (http://moonimpact.eu/umd-package/) av ERC IMPACT-projektet som ett open access-paket.
För att göra det universellt och lättare att hantera, är vårt tillvägagångssätt att först extrahera all information relaterad till det termodynamiska tillståndet och atombanorna från utdatafilen för den faktiska molekylärdynamikkörningen. Den här informationen lagras i en dedikerad fil, vars format är oberoende av det ursprungliga MD-paketet där simuleringen kördes. Vi namnger dessa filer “umd” filer, som står för Universal Molecular Dynamics. På detta sätt kan vårt UMD-paket enkelt användas av alla ab initio-grupper med vilken programvara som helst, allt med en minimal anpassningsinsats. Det enda kravet för att använda det nuvarande paketet är att skriva lämplig toser från utdata från den specifika MD-programvaran till umd-filformatet, om detta ännu inte finns. För närvarande tillhandahåller vi sådana tolkare för VASP8– och QBox9-paketen.
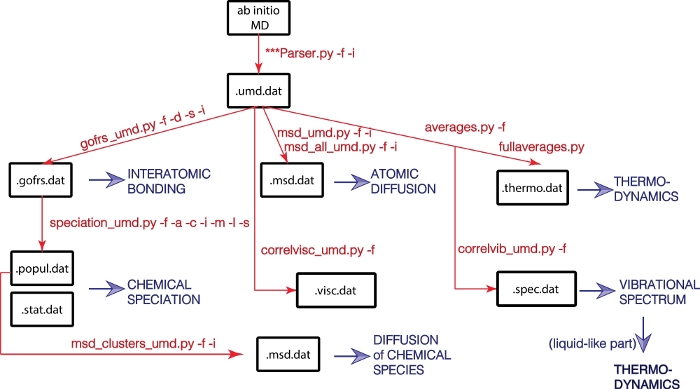
Bild 1: Flödesschema för UMD-biblioteket.
Fysiska egenskaper är i blått och större Python-skript och deras alternativ är i rött. Klicka här för att se en större version av den här figuren.
Umd-filerna är ASCII-filer; typisk förlängning är “umd.dat” men inte obligatorisk. Alla analyskomponenter kan läsa ASCII-filer i umdformatet, oavsett det faktiska namntillägget. Några av de automatiska skript som är utformade för att utföra snabb storskalig statistik över flera simuleringar letar dock specifikt efter filer med umd.dat-tillägget. Varje fysisk egenskap uttrycks på en rad. Varje rad börjar med ett nyckelord. På detta sätt är formatet mycket anpassningsbart och gör det möjligt att läggas till nya egenskaper i umd-filen, samtidigt som dess läsbarhet bevaras i alla versioner. De första 30 raderna i navelfilen för simuleringen av pyrolit vid 4,6 GPa och 3000 K, som används nedan i diskussionen, visas i figur 2.

Figur 2: Början av umdfilen som beskriver simuleringen av flytande pyrolit vid 4,6 GPa och 3000 K.
Rubriken följs av beskrivningen av varje ögonblicksbild. Varje egenskap är skriven på en rad som innehåller namnet på den fysiska egenskapen, värdet eller enheterna och enheterna, alla åtskilda av blanksteg. Klicka här för att se en större version av den här figuren.
Alla umdfiler innehåller en rubrik som beskriver innehållet i simuleringscellen: antalet atomer, elektroner och atomtyper, samt detaljer för varje atom, såsom dess typ, kemiska symbol, antal valenselektroner och dess massa. En tom rad markerar slutet på huvudet och separerar det från huvuddelen av umdfilen.
Sedan beskrivs varje steg i simuleringen. För det första anges de momentana termodynamiska parametrarna, var och en på en annan linje, som anger i) parameterns namn, som energi, påfrestningar, motsvarande hydrostatiskt tryck, densitet, volym, gitterparametrar etc., ii) dess värde och iii) dess enheter. En tabell som beskriver atomerna kommer härnäst. En rubrikrad ger de olika måtten, som kartesiska positioner, hastigheter, avgifter etc., och deras enheter. Sedan beskrivs varje atom på en rad. Av grupper om tre, motsvarande de tre x- y-z-axlarna , är posterna: de reducerade positionerna, de kartesiska positionerna vikta i simuleringscellen, de kartesiska positionerna (som korrekt tar hänsyn till det faktum att atomer kan korsa flera enhetsceller under en simulering), atomhastigheterna och atomkrafterna. De två sista posterna är skalärer: laddning och magnetiskt ögonblick.
Två stora bibliotek säkerställer att hela paketet fungerar korrekt. Det umd_process.py biblioteket behandlar umdfilerna, som att läsa och skriva ut. det crystallography.py biblioteket behandlar all information relaterad till den faktiska atomstrukturen. Den underliggande filosofin i crystallography.py bibliotek är att behandla gitteret som ett vektoriskt utrymme. Enhetscellparametrarna tillsammans med deras orientering representerar basvektorerna. “Rymden” har en serie skalärattribut (specifik volym, densitet, temperatur och specifikt antal atomer), termodynamiska egenskaper (inre energi, tryck, värmekapacitet etc.) och en serie tensoriella egenskaper (stress och elasticitet). Atomer fyller det här utrymmet. Klassen “Lattice” definierar denna ensemble, tillsammans med olika få korta beräkningar, som specifik volym, densitet, erhållande av det ömsesidiga gitteret från det direkta etc. Klassen “Atomer” definierar atomerna. De kännetecknas av en serie skaläregenskaper (namn, symbol, massa, antal elektroner etc.) och en serie vektoregenskaper (positionen i rymden, antingen i förhållande till den vektoriska grunden som beskrivs i Gitterklassen, eller i förhållande till universella kartesiska koordinater, hastigheter, krafter etc.). Bortsett från dessa två klasser innehåller crystallography.py bibliotek en serie funktioner för att utföra en mängd olika tester och beräkningar, till exempel atomavstånd eller cellförökning. Det periodiska systemet för elementen ingår också som en ordlista.
De olika komponenterna i umdpaketet skriver flera utdatafiler. Som en allmän regel är de alla ASCII-filer, alla deras poster separeras av flikar och de görs så självförklarande som möjligt. Till exempel anger de alltid tydligt den fysiska egenskapen och dess enheter. Umd.dat-filerna följer helt denna regel.
