Væsker og smelter er aktive kjemiske og fysiske transportvektorer i naturlige miljøer. De forhøyede ratene av atomdiffusjon favoriserer kjemiske utvekslinger og reaksjoner, den lave viskositeten kombinert med varierende oppdrift favoriserer stor masseoverføring, og krystall-smeltetetthetsrelasjoner favoriserer lagdeling inne i planetlegemer. Fraværet av et periodisk gitter, typiske høye temperaturer som kreves for å nå smeltet tilstand, og vanskeligheten med slukking gjør den eksperimentelle bestemmelsen av en rekke åpenbare egenskaper, som tetthet, diffusjon og viskositet, ekstremt utfordrende. Disse vanskelighetene gjør alternative beregningsmetoder sterke og nyttige verktøy for å undersøke denne klassen av materialer.
Med ankomsten av datakraft og tilgjengeligheten av superdatamaskiner, brukes to store numeriske atomistiske simuleringsteknikker for tiden for å studere den dynamiske tilstanden til et ikke-krystallinsk atomistisk system, Monte Carlo1 og molekylær dynamikk (MD) 1,2. I Monte Carlo-simuleringer prøves konfigurasjonsrommet tilfeldig; Monte Carlo-metoder viser lineær skalering i parallellisering hvis alle prøvetakingsobservasjoner er uavhengige av hverandre. Kvaliteten på resultatene avhenger av kvaliteten på den tilfeldige tallgeneratoren og representativiteten til prøvetakingen. Monte Carlo-metoder viser lineær skalering parallelt hvis prøvetakingen er uavhengig av hverandre. I molekylær dynamikk (MD) prøves konfigurasjonsrommet av tidsavhengige atombaner. Fra en gitt konfigurasjon beregnes atombanene ved å integrere de newtonske bevegelsesligningene. De interatomære kreftene kan beregnes ved hjelp av modellinteratomiske potensialer (i klassisk MD) eller ved hjelp av første-prinsipper metoder (i ab initio, eller første-prinsipper, MD). Kvaliteten på resultatene avhenger av lengden på banen og dens evne til ikke å bli tiltrukket av lokal minima.
Molekylære dynamikksimuleringer inneholder en mengde informasjon, alt relatert til systemets dynamiske oppførsel. Termodynamiske gjennomsnittsegenskaper, som intern energi, temperatur og trykk, er ganske standard for beregning. De kan trekkes ut fra utgangsfilen(e) av simuleringene og gjennomsnittet, mens mengder relatert direkte til bevegelsen av atomene, samt til deres gjensidige forhold, må beregnes etter utvinning av atomposisjoner og hastigheter.
Følgelig har mye arbeid vært dedikert til å visualisere resultatene, og forskjellige pakker er tilgjengelige i dag, på forskjellige plattformer, åpen kildekode eller ikke [Ovito3, VMD4, Vesta5, Travis6, etc.]. Alle disse visualiseringsverktøyene håndterer effektivt med interatomære avstander, og som sådan tillater de effektiv beregning av parfordelingsfunksjoner og diffusjonskoeffisienter. Ulike grupper som utfører simuleringer av storskala molekylær dynamikk har proprietær programvare for å analysere ulike andre egenskaper som følge av simuleringene, noen ganger i shareware eller andre former for begrenset tilgang til samfunnet, og noen ganger begrenset i omfang og bruk til noen spesifikke pakker. Sofistikerte algoritmer for å trekke ut informasjon om interatomær binding, geometriske mønstre og termodynamikk utvikles og implementeres i noen av disse pakkene3,4,5,6,7, etc.
Her foreslår vi UMD-pakken – en åpen kildekode-pakke skrevet i Python for å analysere utgangen av molekylære dynamikksimuleringer. UMD-pakken muliggjør beregning av et bredt spekter av strukturelle, dynamiske og termodynamiske egenskaper (figur 1). Pakken er tilgjengelig via GitHub-nettstedet (https://github.com/rcaracas/UMD_package) og via en dedikert side (http://moonimpact.eu/umd-package/) av ERC IMPACT-prosjektet som en åpen tilgangspakke.
For å gjøre det universelt og enklere å håndtere, er vår tilnærming først å trekke ut all informasjon relatert til den termodynamiske tilstanden og atombanene fra utdatafilen til selve molekylærdynamikkløpet. Denne informasjonen lagres i en dedikert fil, hvis format er uavhengig av den opprinnelige MD-pakken der simuleringen ble kjørt. Vi navngir disse filene “umd” filer, som står for Universal Molecular Dynamics. På denne måten kan UMD-pakken vår enkelt brukes av enhver ab initio-gruppe med hvilken som helst programvare, alt med en minimal innsats for tilpasning. Det eneste kravet for å bruke den nåværende pakken er å skrive riktig parser fra utgangen av den aktuelle MD-programvaren til UMD-filformatet, hvis dette ennå ikke eksisterer. Foreløpig tilbyr vi slike parsere for VASP8– og QBox9-pakkene.
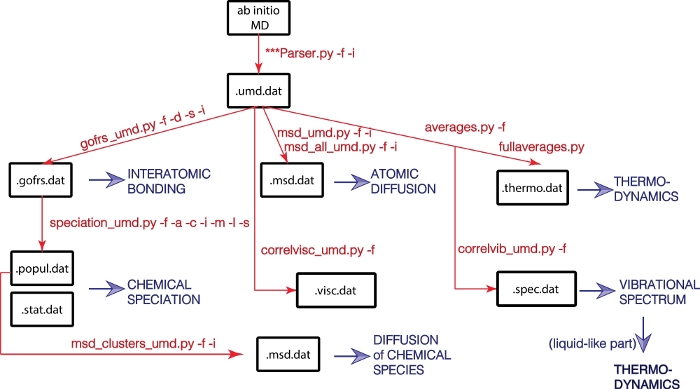
Figur 1: Flytskjema for UMD-biblioteket.
Fysiske egenskaper er i blått, og store Python-skript og deres alternativer er i rødt. Klikk her for å se en større versjon av denne figuren.
Umd-filene er ASCII-filer; typisk utvidelse er “umd.dat”, men ikke obligatorisk. Alle analysekomponentene kan lese ASCII-filer i UMD-formatet, uavhengig av den faktiske navneutvidelsen. Noen av de automatiske skriptene som er utformet for å utføre rask statistikk i stor skala over flere simuleringer, ser imidlertid spesielt etter filer med filtypen UMD.dat. Hver fysiske egenskap uttrykkes på én linje. Hver linje starter med et nøkkelord. På denne måten er formatet svært tilpasningsdyktig og gjør det mulig å legge til nye egenskaper i UMD-filen, samtidig som det bevarer lesbarheten i alle versjoner. De første 30 linjene i umd-filen av simuleringen av pyrolitt ved 4,6 GPa og 3000 K, brukt nedenfor i diskusjonen, vises i figur 2.

Figur 2: Begynnelsen på umd-filen som beskriver simuleringen av flytende pyrolitt ved 4,6 GPa og 3000 K.
Hodet etterfølges av beskrivelsen av hvert øyeblikksbilde. Hver egenskap skrives på én linje, som inneholder navnet på den fysiske egenskapen, verdien(e) og enhetene, alle atskilt med mellomrom. Klikk her for å se en større versjon av denne figuren.
Alle UMD-filer inneholder en overskrift som beskriver innholdet i simuleringscellen: antall atomer, elektroner og atomtyper, samt detaljer for hvert atom, for eksempel type, kjemisk symbol, antall valenselektroner og dens masse. En tom linje markerer slutten på toppteksten og skiller den fra hoveddelen av UMD-filen.
Deretter er hvert trinn i simuleringen detaljert. For det første er de øyeblikkelige termodynamiske parametrene gitt, hver på en annen linje, som spesifiserer (i) navnet på parameteren, som energi, spenninger, tilsvarende hydrostatisk trykk, tetthet, volum, gitterparametere, etc., (ii) dens verdi (er) og (iii) dens enheter. Et bord som beskriver atomene kommer neste gang. En overskriftslinje gir de forskjellige målene, for eksempel kartesiske posisjoner, hastigheter, gebyrer osv., og deres enheter. Deretter er hvert atom detaljert på en linje. Ved grupper på tre, som tilsvarer de tre x-, y-, z-aksene , er oppføringene: de reduserte posisjonene, de kartesiske posisjonene brettet inn i simuleringscellen, de kartesiske posisjonene (som riktig tar hensyn til det faktum at atomer kan krysse flere enhetsceller under en simulering), atomhastighetene og atomkreftene. De to siste oppføringene er skalarer: ladning og magnetisk øyeblikk.
To store biblioteker sikrer at hele pakken fungerer som den skal. Det umd_process.py biblioteket omhandler UMD-filene, som lesing og utskrift. Det crystallography.py biblioteket omhandler all informasjon relatert til den faktiske atomstrukturen. Den underliggende filosofien til det crystallography.py biblioteket er å behandle gitteret som et vektorrom. Enhetscelleparameterne sammen med orienteringen representerer basisvektorene. “Space” har en rekke skalaregenskaper (spesifikt volum, tetthet, temperatur og spesifikt antall atomer), termodynamiske egenskaper (intern energi, trykk, varmekapasitet, etc.), og en rekke tensoriale egenskaper (stress og elastisitet). Atomer fyller dette rommet. “Gitter” -klassen definerer dette ensemblet, sammen med ulike få korte beregninger, som spesifikt volum, tetthet, oppnå det gjensidige gitteret fra den direkte, etc. “Atomer” -klassen definerer atomene. De er preget av en rekke skalaregenskaper (navn, symbol, masse, antall elektroner, etc.) og en rekke vektoregenskaper (posisjonen i rommet, enten i forhold til vektorgrunnlaget beskrevet i Gitterklassen, eller i forhold til universelle kartesiske koordinater, hastigheter, krefter, etc.). Bortsett fra disse to klassene, inneholder det crystallography.py biblioteket en rekke funksjoner for å utføre en rekke tester og beregninger, for eksempel atomavstander eller cellemultiplikasjon. Den periodiske tabellen over elementene er også inkludert som en ordbok.
De ulike komponentene i UMD-pakken skriver flere utdatafiler. Som en generell regel er de alle ASCII-filer, alle oppføringene deres er skilt av faner, og de er laget så selvforklarende som mulig. For eksempel angir de alltid tydelig den fysiske egenskapen og dens enheter. Filene umd.dat er fullstendig i samsvar med denne regelen.
