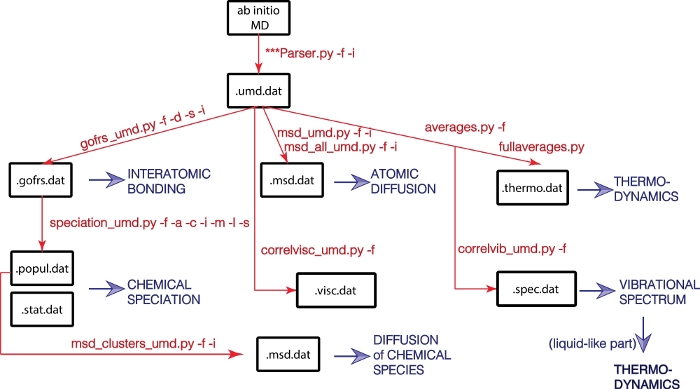
1. Análisis de las corridas de dinámica molecular NOTA: El paquete está disponible a través del sitio web de GitHub (https://github.com/rcaracas/UMD_package) y a través de una página dedicada (http://moonimpact.eu/umd-package/) del proyecto ERC IMPACT como un paquete de acceso abierto. Extraiga cada conjunto específico de propiedades físicas utilizando uno o más scripts de Python dedicados del paquete. Ejecute todos los scripts en la línea de comandos; todos emplean una serie de banderas, que son lo más consistentes posible de un guión a otro. Los indicadores, su significado y los valores predeterminados se resumen en la Tabla 1. Bandera Significado Script usándolo Valor predeterminado -h Ayuda breve todo -f Nombre de archivo UMD todo -i Pasos de termalización a descartar todo 0 -i Archivo de entrada que contiene los enlaces interatómicos especiación bonds.input -s Muestreo de la frecuencia msd, especiación 1 (se considera cada paso) -a Lista de átomos o aniones especiación -c Lista de cationes especiación -l Longitud del enlace especiación 2 -t Temperatura vibraciones, reología -v Discretización de la anchura de la ventana de muestreo de la trayectoria para el análisis de desplazamiento cuadrático medio Msd 20 -z Discretización del inicio de la ventana de muestreo de la trayectoria para el análisis de desplazamiento cuadrático medio Msd 20 Tabla 1: Banderas más comunes utilizadas en el paquete UMD y su significado más común. Comience transformando la salida de la simulación MD realizada en un código de primeros principios, como VASP8 o QBox9, en un archivo UMD. Si las simulaciones de MD se realizaron en VASP, entonces en la línea de comandos escriba:VaspParser.py -f -i donde el indicador –f define el nombre del archivo VASP OUTCAR y el –i la longitud de termalización.NOTA: El paso inicial, definido por –i permite descartar los primeros pasos de las simulaciones, que representan la termalización. En una ejecución típica de dinámica molecular, la primera parte del cálculo representa la termalización, es decir, el tiempo que tarda el sistema en describir una distribución gaussiana de la temperatura, y en que todo el sistema exhiba fluctuaciones de la temperatura, presión, energía, etc. alrededor de los valores de equilibrio. Esta parte de termalización de la simulación no debe tenerse en cuenta al analizar las propiedades estadísticas del fluido. Transforme el archivo . umd archivos en . xyz para facilitar la visualización en varios otros paquetes, como VMD4 o Vesta5. En la línea de comandos, escriba:umd2xyz.py -f -i -s donde –f define el nombre del . umd file, –i define el período de termalización a descartar, y –s la frecuencia del muestreo de la trayectoria almacenada en el . archivo umd . Los valores predeterminados son –i 0 –s 1, es decir, considerando todos los pasos de la simulación, sin descartar ninguno. Invierta el archivo umd en archivos POSCAR de tipo VASP utilizando el script umd2poscar.py; las instantáneas de las simulaciones se pueden seleccionar con una frecuencia predefinida. En la línea de comandos, escriba:umd2poscar.py -f -i -l -s donde –l representa el último paso que se va a transformar en archivo POSCAR. Los valores predeterminados son -i 0 -l 10000000 -s 1. Este valor de –l es lo suficientemente grande como para cubrir una trayectoria completa típica. 2. Realizar el análisis estructural Ejecute el script gofrs_umd.py para calcular la función de distribución de pares (PDF) gᴀʙ(r) para todos los pares de tipos atómicos A y B (Figura 3). La salida se escribe en un archivo ASCII, separado por tabulaciones, con la extensión gofrs.dat. En la línea de comandos, escriba:gofrs_umd.py -f -s -d -i NOTA: Los valores predeterminados son Sampling_Frequency (la frecuencia para muestrear la trayectoria) = 1 paso; DiscretizationInterval (para trazar el g(r)) = 0,01 Å; InitialStep (número de pasos al principio de la trayectoria que se descartan) = 0. El PDF radial, gᴀʙ(r) es el número medio de átomos de tipo B a una distancia d_ᴀʙ dentro de una capa esférica de radio r y espesor dr centrada en los átomos de tipo A (Figura 3):con ρ la densidad atómica, NA y NB el número de átomos de tipo A y B, y δ(r−rᴀʙ) la función delta que es igual a 1 si los átomos A y B se encuentran a una distancia entre r y r+dr. La abscisa del primer máximo de gᴀʙ(r) da la longitud de enlace de mayor probabilidad entre los átomos de tipo A y B, que es la más cercana a una distancia de enlace promedio que podemos determinar. El primer mínimo delimita el alcance de la primera esfera de coordinación. Por lo tanto, la integral sobre el PDF hasta el primer mínimo da el número de coordinación promedio. La suma de las transformadas de Fourier de gᴀʙ(r) para todos los pares de tipos atómicos A y B produce el patrón de difracción del fluido, tal como se obtiene experimentalmente con un difractómetro. Sin embargo, en realidad, como a menudo las esferas de coordinación de alto orden faltan en el gᴀʙ(r), el patrón de difracción no se puede obtener en su totalidad. Figura 3: Determinación de la función de distribución de pares.a) Para cada átomo de una especie (por ejemplo, rojo), todos los átomos de la especie coordinadora (por ejemplo, gris y/o rojo) se cuentan en función de la distancia. (b) El gráfico de distribución de distancia resultante para cada instantánea, que en esta etapa es solo una colección de funciones delta, se promedia sobre todos los átomos y todas las instantáneas y se pondera mediante la distribución de gas ideal para generar (c) la función de distribución de pares que es continua. El primer mínimo de g(r) es el radio de la primera esfera de coordinación, utilizado posteriormente en el análisis de especiación. Haga clic aquí para ver una versión más grande de esta figura. Extraer las distancias medias de enlace interatómico como los radios de las primeras esferas de coordinación. Para ello, identifique la posición del primer máximo de las funciones gᴀʙ(r): trace el archivo gofrs.dat en una aplicación de hoja de cálculo y busque los máximos y mínimos para cada par de átomos. Identifique el radio de la primera esfera de coordinación, como el primer mínimo del PDF, gᴀʙ(r), utilizando un software de hoja de cálculo. Esta es la base para todo el análisis estructural del fluido; el PDF produce el estado de enlace promedio de los átomos en el fluido. Extraiga las distancias de los primeros mínimos, es decir, la abscisa, y escríbalas en un archivo separado, llamado, por ejemplo, bonds.input. Alternativamente, ejecute uno de los scripts analyze_gofr del paquete UMD para identificar los máximos y los mínimos de las funciones gᴀʙ(r). En la línea de comandos, escriba:analyze_gofr_semi_automatic.py Haga clic en la posición del máximo y el mínimo de la función gᴀʙ(r) que se muestra en el gráfico que abre el programa. El script escanea automáticamente la carpeta actual, identifica todos los archivos gofrs.dat y realiza el análisis para cada uno de ellos. Haga clic de nuevo en el máximo y el mínimo en la ventana cada vez que el script necesite una suposición inicial informada. Abra y mire el archivo generado automáticamente llamado bonds.input que contiene las distancias de enlace interatómico. 3. Realizar el análisis de especiación Calcular la topología de enlace entre los átomos, utilizando el concepto de conectividad dentro de la teoría de grafos: los átomos son los nodos y los enlaces interatómicos son los caminos. El script speciation_umd.py necesita las distancias de enlace interatómico definidas en el archivo bonds.input .NOTA: La matriz de conectividad se construye en cada paso temporal: dos átomos que se encuentran a una distancia menor que el radio de su correspondiente primera esfera de coordinación se consideran unidos, es decir, conectados. Varias redes atómicas se construyen tratando los átomos como nodos en un gráfico cuyas conexiones están definidas por este criterio geométrico. Estas redes son las especies atómicas, y su conjunto define la especiación atómica en ese fluido en particular (Figura 4). Figura 4: Identificación de los cúmulos atómicos.Los poliedros de coordinación se definen utilizando distancias interatómicas. Todos los átomos a una distancia menor que un radio especificado se consideran enlazados. Aquí el umbral corresponde a la primera esfera de coordinación (los círculos rojos claros), definida en la Figura 1. La polimerización y, por lo tanto, las especies químicas se obtienen de las redes de los átomos unidos. Nótese el cúmulo central Red1Grey2, que está aislado de los otros átomos, que forman un polímero infinito. Haga clic aquí para ver una versión más grande de esta figura. Ejecute el script de especiación para obtener la matriz de conectividad y obtener los poliedros de coordinación o la polimerización. En la línea de comandos, escriba:speciation_umd.py -f -s -i -l -c -a -m -r donde el indicador -i da el archivo con las distancias de enlace interatómico, que se produjo por ejemplo en el paso anterior. Alternativamente, ejecute el script con una sola longitud para todos los enlaces definidos por el indicador -l.NOTA: La bandera -c especifica los átomos centrales, y la bandera -a los ligandos. Tanto los átomos centrales como los ligandos pueden ser de diferentes tipos; en este caso, deben estar separados por comas. La bandera -m da el tiempo mínimo que una especie debe vivir para ser considerada en el análisis. De forma predeterminada, este tiempo mínimo es cero, todas las ocurrencias se cuentan en el análisis final. Ejecute el script speciation_umd.py con el indicador –r 0, que muestra el gráfico de conectividad en el primer nivel para identificar los poliedros de coordinación. Por ejemplo, un átomo central, denotado como un catión , puede estar rodeado por uno o más aniones (Figura 4). La secuencia de comandos de especiación identifica cada uno de los poliedros de coordinación. La media ponderada de todos los poliedros de coordinación da el número de coordinación, idéntico al obtenido de la integración del PDF. En la línea de comandos, escriba:speciation_umd.py -f -i -c -a -r 0NOTA: Los números de coordinación promedio en fluidos son números fraccionarios. Esta fraccionalidad proviene de la característica media de la coordinación. La definición basada en la especiación produce una representación más intuitiva e informativa de la estructura del fluido, donde se cuantifican las proporciones relativas de las diferentes especies, es decir, las coordinaciones. Ejecute el script speciation_umd.py con el indicador –r 1, que muestrea el gráfico de conectividad en todos los niveles de profundidad para obtener la polimerización. La red a través del gráfico atómico tiene una cierta profundidad, ya que los átomos están unidos más lejos a otros enlaces (por ejemplo, en secuencias de cationes y aniones alternos) (Figura 4). Abra los dos archivos . popul.dat y . stat.dat consecutivamente; estos constituyen la salida del script de especiación. Cada cúmulo está escrito en una línea, especificando su fórmula química, el momento en que se formó, el momento en que murió, su vida útil, una matriz con la lista de los átomos que forman este cúmulo. Trazar la vida útil de cada cúmulo atómico de todas las especies químicas encontradas en la simulación como se encuentra en el archivo .popul.dat (Figura 5). Trazar el análisis poblacional con la abundancia de cada especie, tal y como se encuentra en el . archivo stat.dat . Este análisis, tanto absoluto como relativo, corresponde a las estadísticas reales de los poliedros de coordinación para el caso -r 0; para el caso de la polimerización, con -r 1 esto debe tratarse cuidadosamente, ya que podría ser necesario aplicar cierta normalización sobre el número relativo de átomos. La abundancia corresponde a la integral a lo largo de las vidas. El. stat.dat archivo también enumera el tamaño de cada clúster, es decir, cuántos átomos lo forman. 4. Calcular los coeficientes de difusión Extraer los desplazamientos cuadráticos medios (MSD) de los átomos en función del tiempo para obtener la autodifusividad. La fórmula estándar del MSD es:donde los prefactores son renormalizaciones. Con la herramienta MSD, hay diferentes formas de analizar los aspectos dinámicos de los fluidos.NOTA: T es el tiempo total de la simulación y N α es el número de átomos de tipo α. El tiempo inicial t0 es arbitrario y abarca la primera mitad de la simulación. Ninit es el número de veces iniciales. τ es la anchura del intervalo de tiempo durante el cual se calculan los TME; su valor máximo es la mitad de la duración de la simulación. En las implementaciones típicas de MSD, cada ventana comienza al final de la anterior. Pero un muestreo más escaso puede acelerar el cálculo del MSD, sin alterar la pendiente del MSD. Para esto, la ventana i-ésima comienza en el tiempo t0(i), pero la ventana (i+1)-ésima comienza en el tiempo t0(i) + τ + v, donde el valor de v está definido por el usuario. Del mismo modo, el ancho de la ventana se incrementa en pasos discretos definidos por el usuario, como tales: τ(i) = τ(i-1) + z. Los valores de z (“paso horizontal”) y v (“paso vertical”) son positivos o cero; el valor predeterminado para ambos es 20. Calcule el MSD utilizando la serie de scripts de msd_umd . Su salida se imprime en un archivo . msd.dat archivo, donde el MSD de cada tipo atómico, átomo o clúster se imprime en una columna en función del tiempo. Calcule el MSD promedio de cada tipo atómico. Los MSD se calculan para cada átomo y luego se promedian para cada tipo atómico. El archivo de salida contiene una columna para cada tipo atómico. En la línea de comandos, escriba:msd_umd.py -f -z -v -b Calcular el MSD de cada átomo. Los MSD se calculan para cada átomo y luego se promedian para cada tipo atómico. El archivo de salida contiene una columna para cada átomo en la simulación y, a continuación, una columna para cada tipo atómico. Esta característica permite identificar átomos que se difunden en dos entornos diferentes, como líquido y gas, o dos líquidos. En la línea de comandos, escriba:msd_all_umd.py -f -z -v -b Calcular el MSD de la especie química. Utilice la población de clústeres identificados con el script de especiación e impresos en el archivo . popul.dat archivo. Los MSD se calculan para cada clúster individual. El archivo de salida contiene una columna para cada clúster. Para evitar considerar polímeros a gran escala, ponga un límite en el tamaño del clúster; su valor predeterminado es 20 átomos. En la línea de comandos, escriba:msd_cluster_umd.py -f -p -s -b -c NOTA: Los valores predeterminados son: –b 100 –s 1 –c 20. Traza el MSD usando un software basado en hojas de cálculo (Figura 6). En una representación log-log del MSD versus el tiempo, identifique el cambio de pendiente. Separe la primera parte, generalmente corta, que representa el régimen balístico , es decir, la conservación de la velocidad de los átomos después de las colisiones. La segunda parte más larga representa el régimen difusivo , es decir, la dispersión de la velocidad de los átomos después de las colisiones. Calcule los coeficientes de difusión a partir de la pendiente del MSD como:donde Z es el número de grados de libertad (Z = 2 para la difusión en el plano, Z = 3 para la difusión en el espacio), y t es el paso del tiempo. 5. Funciones de correlación de tiempo Calcule las funciones de correlación de tiempo como una medida de la inercia del sistema utilizando la fórmula general:A puede ser una variedad de variables dependientes del tiempo, como las posiciones atómicas, las velocidades atómicas, las tensiones, la polarización, etc., cada una de las cuales produce, a través de las relaciones de Green-Kubo12,13, diferentes propiedades físicas, a veces después de una transformación adicional. Analizar las velocidades atómicas para obtener el espectro vibratorio del líquido y la expresión alternativa de los coeficientes de autodifusión atómica. Ejecute el script vibr_spectrum_umd.py para calcular la función de autocorrelación velocidad atómica (VAC) para cada tipo atómico y realice su transformada de Fourier rápida. En la línea de comandos, escriba:vibr_spectrum_umd.py -f -t donde –t es la temperatura que debe definir el usuario. El script imprime dos archivos: el archivo . vels.scf.dat archivo con la función VAC para cada tipo atómico y el archivo . vibr.dat archivo con el espectro vibratorio descompuesto en cada especie atómica y el valor total. Abra y lea el .dat vels.scf. Trace la función VAC desde el archivo vels.scf.dat utilizando un software similar a una hoja de cálculo. Mantenga la parte real del Fourier VAC. Esto es lo que produce el espectro vibratorio, en función de la frecuencia:donde m son las masas atómicas. Traza el espectro vibratorio desde el archivo vibr.dat utilizando un software similar a una hoja de cálculo (Figura 7). Identificar el valor finito en ω=0 que corresponde al carácter difusivo del fluido y los diversos picos del espectro a frecuencia finita. Identificar la participación de cada tipo atómico en el espectro vibratorio.NOTA: La descomposición en tipos atómicos muestra que diferentes átomos tienen diferentes contribuciones ω=0, correspondientes a sus coeficientes de difusión. La forma general del espectro es mucho más suave con menos características que para un sólido correspondiente. En la cáscara, lea la integral sobre el espectro vibratorio, que produce los coeficientes de difusión para cada especie atómica.NOTA: Las propiedades termodinámicas se pueden obtener mediante la integración del espectro vibratorio, pero los resultados deben usarse con precaución debido a dos aproximaciones: la integración es válida dentro de la aproximación cuasi armónica, que no necesariamente se mantiene a altas temperaturas; y es necesario descartar la parte gaseosa del espectro correspondiente a la difusión. La integración debe hacerse solo sobre la parte del espectro similar a una red. Pero esta separación generalmente requiere varios pasos y cálculos posteriores al procesamiento14, que no están cubiertos por el presente paquete UMD. Ejecute el script viscosity_umd.py para analizar la autocorrelación del tensor de tensión de los componentes para estimar la viscosidad de la masa fundida. En la línea de comandos, escriba:viscosity_umd.py -f -i -s -o -l NOTA: Esta característica es exploratoria y cualquier resultado debe tomarse con precaución. En primer lugar, comprobar a fondo la convergencia de la viscosidad con respecto a la longitud de la simulación. Derivar la viscosidad del fluido a partir de la autocorrelación del tensor de tensión15 como:donde V y T son el volumen y la temperatura respectivamente, κB es la constante de Boltzmann y σ ij la componente ij fuera de diagonal del tensor de tensión, expresada en coordenadas cartesianas. Utilizar un ajuste más adecuado para obtener una estimación más robusta de la viscosidad15,16 y evitar el ruido de la función de autocorrelación tensor-tensión que pudiera derivarse del tamaño finito y la duración finita de las simulaciones. Para la función de autocorrelación del tensor de tensión, utilice la siguiente forma funcional15,16 que produzca buenos resultados:donde A, B, τ1, τ2 y ω son parámetros de ajuste. Después de la integración, la expresión para la viscosidad se convierte en: 6. Parámetros termodinámicos derivados de las simulaciones. Ejecute averages.py para extraer los valores promedio y la propagación (como desviación estándar) de la presión, la temperatura, la densidad y la energía interna de los archivos umd . En la línea de comandos, escriba:averages.py -f -s con –s 0 como valor predeterminado. Calcular el error estadístico del promedio, utilizando métodos de bloqueo.NOTA: Hay varios sabores de este método. Siguiendo el trabajo de Allen y Tildesley2, es común promediar sobre secuencias de bloques temporales, de longitud cada vez mayor, y estimar la desviación estándar con respecto a la media aritmética17. La convergencia puede alcanzarse en el límite de muchos tamaños de bloque y lo suficientemente largos, cuando el muestreo no está correlacionado. Aunque el valor umbral real para la convergencia generalmente debe elegirse manualmente. Utilice el método de reducción a la mitad18: a partir de la muestra de datos inicial, en cada paso κ, reduzca a la mitad el número de muestras promediando cada dos muestras consecutivas correspondientes del paso anterior κ−1: Ejecute el script fullaverages.py para realizar el análisis estadístico completo, incluido el error de la media. En la línea de comandos, escriba:fullaverages.py -s -u NOTA: El script se automatiza hasta el punto de buscar todos los archivos .umd.dat en el directorio actual y realizar el análisis para todos ellos. Los valores predeterminados son –s 0 –u 0. Para -u 0 la salida es mínima, y para -u 1 la salida es completa, con varias unidades alternativas impresas. Este script requiere soporte gráfico, ya que crea una imagen gráfica para comprobar la convergencia para estimar el error en la media.