Preliminary qRT-PCR data suggested that an EWS/FLI mutant called DAF, with specific tyrosine to alanine mutations in the repetitive and disordered region of EWS, maintained the ability to activate EWS/FLI target genes, but failed to repress critical target genes23. In order to better understand the relationship between these residues in the EWS domain and EWS/FLI function, the protocol described above and outlined in Figure 1 was used. A673 Ewing sarcoma cells were virally transduced with an shRNA targeting the 3’UTR of FLI1, resulting in the depletion of endogenous EWS/FLI. After four days of selection, EWS/FLI function was rescued with viral transduction of different 3XFLAG-tagged EWS/FLI mutant constructs, with empty vector as a control for no rescue. A non-functional mutant lacking the EWS domain, called Δ22, was used as a negative control and wild-type EWS/FLI, called wtEF, was used as a positive control (Figure 2A). DAF was used as the test construct, though more than one test construct can be used if desired. Cells were selected for an additional 10 days to allow construct expression to stabilize and then collected for RNA (with a gDNA removal step), protein and colony forming assays. Four replicates were collected and representative qRT-PCR and western blots showing effective knockdown and rescue are shown in Figure 2B-D. It should be noted that DAF-rescued cells failed to form colonies as shown in Figure 2E, suggesting impaired oncogenic transformation.
Following completion of the replicate validation and phenotypic assays, RNA was submitted to the Institute for Genomic Medicine at Nationwide Children’s Hospital for library preparation and next generation sequencing with ~50 million 150-bp paired-end reads collected. The data was returned as fastq.gz files. Low-quality reads were trimmed from these files with TrimGalore and STAR was used to align reads to the human genome hg19 and count the reads per gene. hg19 was used for purposes of compatibility with the other curated datasets for EWS/FLI used in downstream analysis. These read counts were combined into a single count matrix for all samples, the first 6 rows of which are shown in Figure 3.
Counts were initially run through DESeq2 without batch normalization, however, visual inspection of the sample-to-sample distance showed potential confounding batch effects as shown highlighted with red arrows in Figure 4A. This likely arose due to biological variability introduced by the passage of cells in culture and differences in the processing of each batch. Normalization for batch effects was performed with ComBat and is generally recommended. The sample-to-sample distances of the batch-normalized data are shown in Figure 4B. Following batch normalization, DESeq2 was used to generate transcriptional profiles for the three constructs (wtEF, Δ22, and DAF) relative to the baseline. Note that while “parental” A673 cells (mock knockdown and mock rescue, called “iLuc” here) were included in the differential analysis, the reference for this experiment are the cells with EWS/FLI-depleted, called iEF cells. The transcriptional profile can be generated for the endogenous protein here by comparing the iLuc sample to iEF, and this may be of interest in understanding how the rescue system works, but that is not the goal of this particular analysis. The transcriptional profiles generated for the mutants include positive (wtEF) and negative (Δ22) controls, with respect to iEF, such that these should function as the benchmarks for other mutants. This is important, as the positive control in this example did not completely recapitulate the function of endogenous EWS/FLI as discussed elsewhere7,23.
The principal component analysis (PCA) in Figure 5 suggests that the transcriptional profile of DAF is intermediate between wtEF and Δ22, confirming partial function. Moreover, hierarchical clustering of the 1000 most variable genes across samples showed that DAF failed to repress EWS/FLI target genes, and only partially retained gene activation activity as shown in Figure 6A and Figure S5. ToppGene analysis suggested that the classes of genes that DAF activates are functionally distinct from those EWS/FLI-activated targets where DAF is non-functional (Figure 6B). Interestingly, the function of activated genes rescued by wtEF, but not by DAF, appear to be related to transcriptional control and chromatin regulation. Based on the results of the colony formation assays, the genes from this core gene signature should be further analyzed for their role in EWS/FLI-mediated oncogenesis. The importance of EWS/FLI-mediated gene repression has been previously described17.
It is known that EWS/FLI possesses a unique binding affinity for GGAA-microsatellite repeat elements19,22, and that binding at these elements drives downstream gene regulation11,15,18,20,22. These microsatellites have been characterized as either associated with activation or repression, and either proximal to (< 5 kb) TSS or distal to (> 5 kb) TSS25. In addition, there are EWS/FLI-regulated genes with high affinity (HA) ETS motifs proximal to TSS23. In order to further analyze the characteristics of DAF function and what types of EWS/FLI-activated genes DAF was able to rescue, differential expression of genes associated with these different classes was analyzed. Interestingly, DAF was most able to rescue GGAA-microsatellite activated genes, but unable to rescue activated genes near an HA site as seen in Figure 7. As seen with hierarchical clustering, DAF fails to rescue EWS/FLI-mediated repression across motif classes. These data suggest that DAF retains sufficient structural features of EWS to bind to and activate from GGAA-microsatellites, both proximal and distal to TSS. This likely arises from the intact SYGQ domain thought to be important for EWS/FLI activity at GGAA repeats11. These data also suggest that the specific tyrosines mutated in DAF play important, but poorly understood, roles in EWS/FLI-mediated gene regulation from HA sites, as well as in gene repression, highlighting an important area of further investigation.
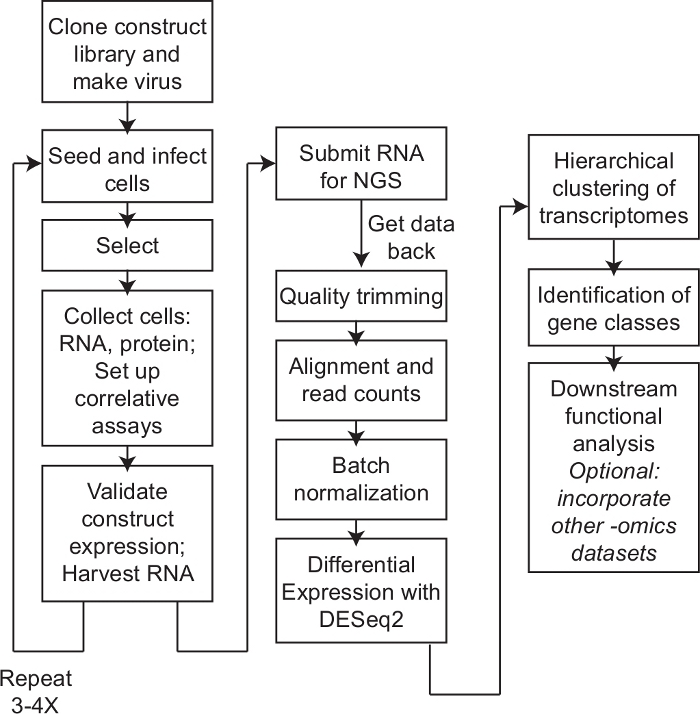
Figure 1: Workflow. Depiction of the step-by-step procedure to perform structure-function mapping by transcriptomics. Cells were first prepared to express the suite of constructs required for structure-function mapping. Following expression, cells were harvested for RNA and protein and assayed for correlative phenotypes. Expression of the constructs was validated, and this process was repeated 3-4 times to gather independent biological replicates. RNA was then submitted for next-generation sequencing (NGS). When data was received, data was trimmed for quality, aligned, and counts per transcript were calculated. Batch effects were controlled for and transcriptomic signatures and differential expression were determined using DESeq2. Hierarchical clustering and downstream analysis integrating other -omics datasets and different pathway or functional analysis can be incorporated. Please click here to view a larger version of this figure.
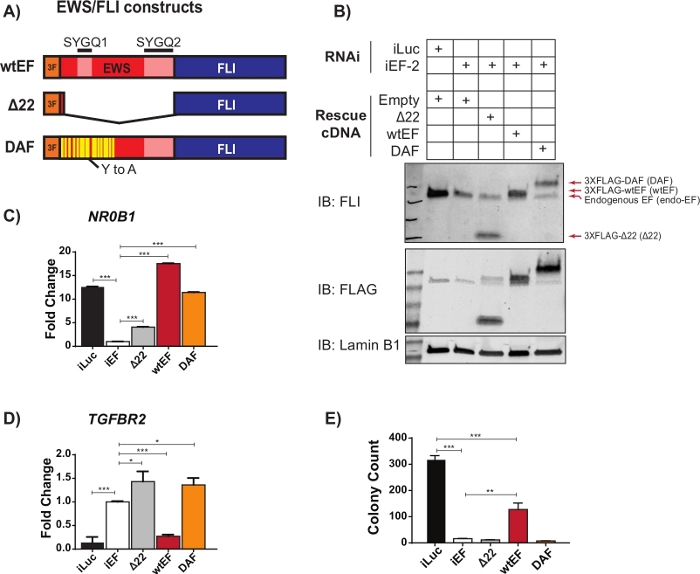
Figure 2: Validation of construct expression and correlative assays. (A) Schematic depicting the constructs tested in this example. (B) Validation of knockdown of endogenous EWS/FLI and expression of 3X-FLAG-tagged constructs by immunoblot. (C,D) Validation of construct activity at an EWS/FLI (C) activated target gene, NR0B1, and (D) repressed target gene, TGFBR2, by qRT-PCR. Data are presented as mean +/- standard deviation. P-values were calculated with a Tukey’s honest significance test. * p < 0.05, ** p < 0.01, *** p < 0.005 (E) Colony counts from soft-agar assays performed to assess transforming activity of constructs. P-values were calculated with a Tukey’s honest significance test. * p < 0.05, ** p < 0.01, *** p < 0.005. This figure is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.

Figure 3: Final collated count data for analysis. Screenshot of the first 6 rows of the count file with gene counts for all the samples to be batch normalized and analyzed. Please click here to view a larger version of this figure.
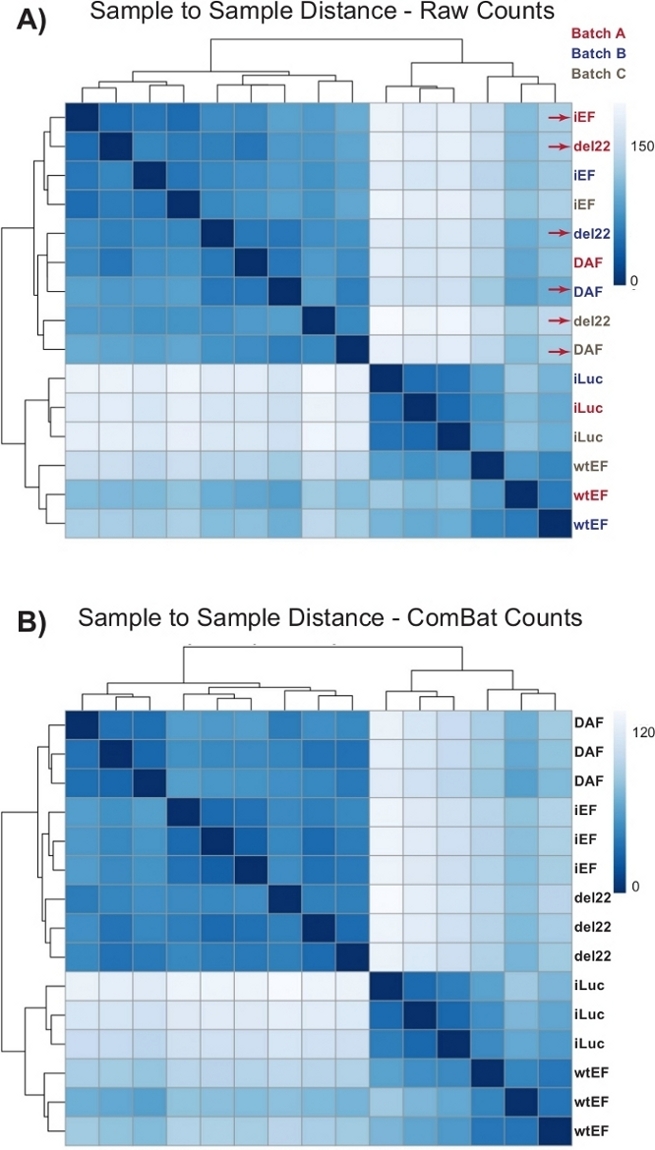
Figure 4: Sample-to-sample distance heatmaps. (A) Sample-to-sample distance plot showing the sample clustering of the raw count data. Samples which are clustering both by batch and by sample are denoted with red arrows. (B) Sample-to-sample distance plot following batch normalization with ComBat. Here, samples from all replicates cluster together, independent of batch. Please click here to view a larger version of this figure.
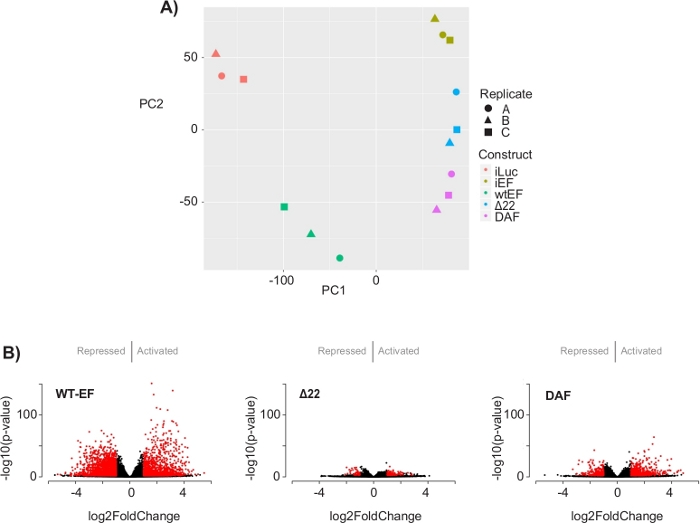
Figure 5: Results of differential expression analysis. (A) Principle component analysis (PCA) plot of the transcriptomic signatures generated for all the samples show strong intra-sample clustering and demonstrate that DAF is intermediated between the positive (wtEF) and negative (Δ22) controls. (B) Volcano plots showing the -log(p-value) plotted against the log2FoldChange for genes in each construct. Genes with an adjusted p-value < 0.05 and a |log2(FoldChange)| > 1 are considered significant and are shown in red. Panel 5B is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.
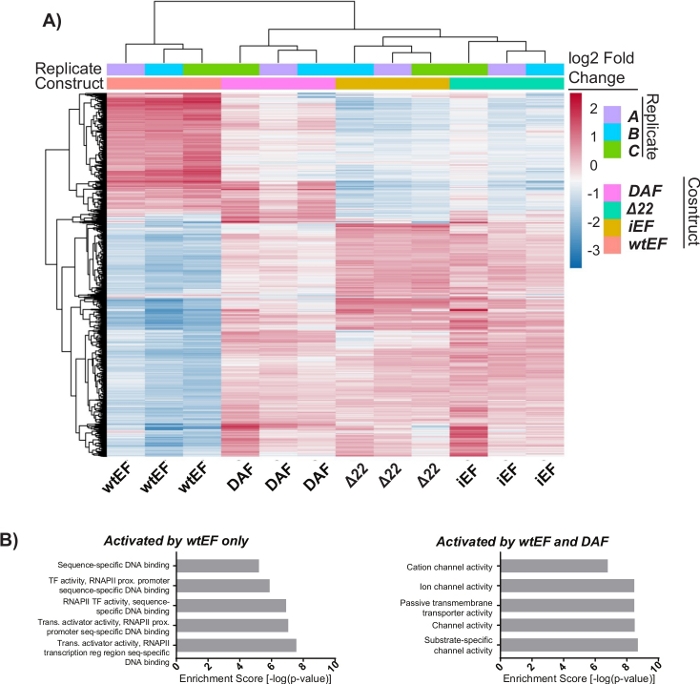
Figure 6: Hierarchical clustering to identify gene classes. (A) Hierarchical clustering of the top 1000 most variable genes across all constructs and the baseline, iEF, shows DAF partially rescues EWS/FLI-mediated gene activation. (B) Gene ontology (molecular function) results from ToppGene showing the functional enrichment of EWS/FLI-activated genes that are either rescued or not rescued by DAF. Panel 6B is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.
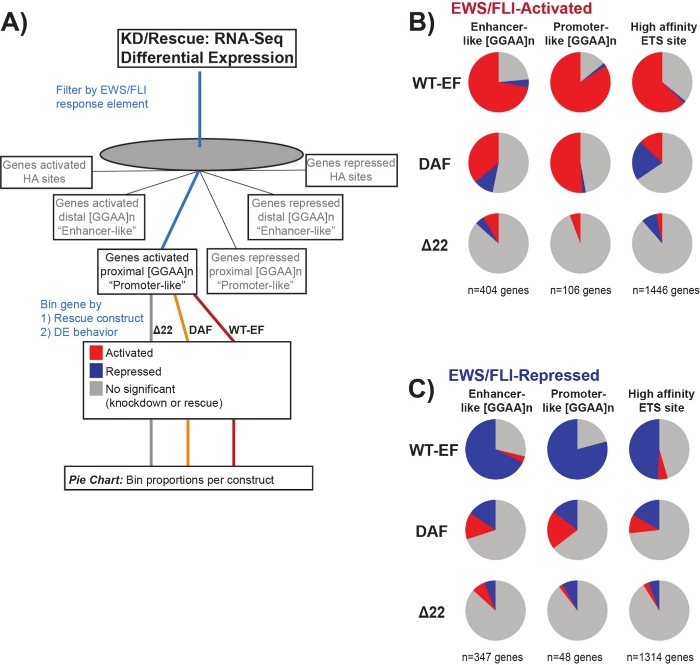
Figure 7: Detailed analysis of different transcription factor response elements to different constructs: (A) Schematic depicting the data processing used to generate panels (B) and (C) by incorporating other available datasets with the transcriptomic profiles here. (B,C) Compilation showing the rescue of different classes of direct EWS/FLI- (B) activated and (C) repressed targets. Genes included were only those genes with detectable differential expression by endogenous EWS/FLI. In each pie chart, gray depicts the portion of genes which are not rescued by the construct. Red depicts the portion of genes that are differentially activated, and blue depicts the portion of genes that are differentially repressed. This figure is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.
Figure S1: Loading the fastq.gz files to the HPC environment, trimming and alignment. Please click here to download this figure.
Figure S2: Collating read counts across samples and running batch normalization with ComBat. Please click here to download this figure.
Figure S3: Running DESeq2 and extracting results of differential expression analysis. Please click here to download this figure.
Figure S4: Analyzing output. Please click here to download this figure.
Figure S5: Hierarchical clustering to identify gene classes: Hierarchical clustering of the top 1000 most variable genes across all constructs and the baseline, iEF, sorted into k clusters. In this instance k=7, but this parameter is set by the user as shown in Figure S4D. Please click here to download this figure.
Table S1: List of genes (Ensembl gene ID) with cluster annotation. Please click here to download this table.
