RNA-seq широко использовался на протяжении многих лет, как правило, для оценки дифференциальной экспрессии генов и открытия генов1. Кроме того, он также может быть использован для оценки различного использования на уровне экзонов из-за генов, экспрессирующих различные изоформы, что способствует лучшему пониманию регуляции генов на посттранскрипционном уровне. Большинство эукариотических генов генерируют различные изоформы путем альтернативного сплайсинга (AS) для увеличения разнообразия экспрессии мРНК. События AS можно разделить на различные паттерны: пропуск полных экзонов (SE), где («кассетный») экзон полностью удаляется из стенограммы вместе с его фланкирующими интронами; альтернативный (донорский) 5-футовый выбор места сращивания (A5SS) и альтернативный 3-дюймовый (акцепторный) выбор места сращивания (A3SS), когда два или более участков сращивания присутствуют на обоих концах экзона; удержание интронов (RI), когда интрон сохраняется в зрелом транскрипте мРНК, и взаимное исключение использования экзона (MXE), где только один из двух доступных экзонов может быть сохранен за один раз 2,3. Альтернативное полиаденилирование (АПА) также играет важную роль в регулировании экспрессии генов с использованием альтернативных поли(А) сайтов для генерации нескольких изоформ мРНК из одного транскрипта4. Большинство участков полиаденилирования (pAs) расположены в 3′ нетранслируемой области (3′ UTR), генерируя изоформы мРНК с различными длинами UTR 3′. Поскольку 3′ UTR является центральным узлом для распознавания регуляторных элементов, различные длины 3′ UTR могут влиять на локализацию, стабильность и трансляцию мРНК5. Существует класс 3′-концевых анализов секвенирования, оптимизированных для обнаружения APA, которые отличаются деталями протокола6. Конвейер, описанный здесь, предназначен для PolyA-seq, но может быть адаптирован для других протоколов, как описано.
В данном исследовании мы представляем конвейер методов дифференциального анализа экзонов 7,8 (рисунок 1), которые можно разделить на две широкие категории: основанные на экзонах (DEXSeq9, diffSplice10) и событийные (реплицированный многомерный анализ сплайсинга транскриптов (rMATS)11). Методы, основанные на экзонах, сравнивают изменение складки в разных условиях отдельных экзонов с мерой общего изменения складки генов, чтобы вызвать дифференциально экспрессированное использование экзонов, и на основе этого вычисляют меру активности АС на уровне гена. Методы, основанные на событиях, используют считывание переходов, охватывающих экзон-интрон, для обнаружения и классификации конкретных событий сплайсинга, таких как пропуск экзона или удержание интронов, и различают эти типы AS в выходных данных3. Таким образом, эти методы обеспечивают взаимодополняющие взгляды для полного анализа AS 12,13. Мы выбрали DEXSeq (на основе пакета DESeq214 DGE) и diffSplice (на основе пакета Limma10 DGE) для исследования, поскольку они являются одними из наиболее широко используемых пакетов для дифференциального анализа сплайсинга. rMATS был выбран в качестве популярного метода для анализа на основе событий. Другим популярным методом, основанным на событиях, является MISO (Смесь изоформ)1. Для APA мы адаптируем подход, основанный на экзонах.
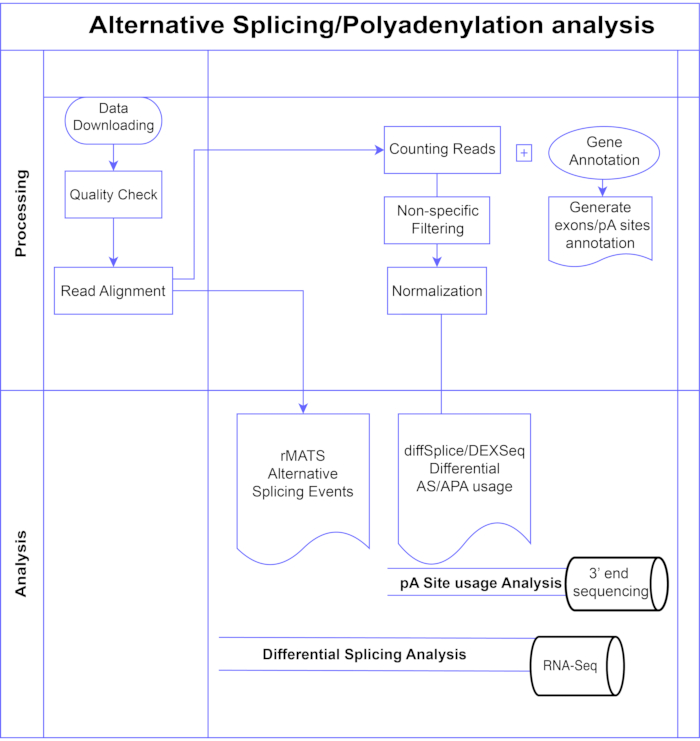
Рисунок 1. Конвейер анализа. Блок-схема шагов, используемых в анализе. Шаги включают в себя: получение данных, выполнение проверок качества и выравнивания считывания с последующим подсчетом считываний с использованием аннотаций для известных экзонов, интронов и участков pA, фильтрацию для удаления низких значений и нормализации. Данные PolyA-seq были проанализированы для альтернативных участков pA с использованием методов diffSplice/DEXSeq, объемная РНК-Seq была проанализирована для альтернативного сплайсинга на уровне экзона методами diffSplice/DEXseq, а события AS анализировались с помощью rMATS. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
Данные RNA-seq, использованные в этом исследовании, были получены из Gene Expression Omnibus (GEO) (GSE138691)15. Мы использовали данные RNA-seq мышей из этого исследования с двумя группами состояний: дикий тип (WT) и Muscleblind-подобный нокаут типа 1 (Mbnl1 KO) с тремя репликами в каждой. Чтобы продемонстрировать дифференциальный анализ использования участка полиаденилирования, мы получили данные PolyA-seq о фибробластах эмбриона мыши (MEFs) (GEO Accession GSE60487)16. Данные имеют четыре группы условий: Wild-type (WT), Muscleblind-like type1/type 2 double knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO с Mbnl3 knockdown (KD) и Mbnl1/2 DKO с управлением Mbnl3 (Ctrl). Каждая группа условий состоит из двух реплик.
| Присоединение к ГЭП | Номер запуска SRA | Пример имени | Состояние | Повторять | Ткань | Секвенирование | Длина чтения | |
| РНК-Сек | GSM4116218 | СРР10261601 | Mbnl1KO_Thymus_1 | Нокаут Mbnl1 | Представитель 1 | Тимус | Сопряженный конец | 100 бит/с |
| GSM4116219 | СРР10261602 | Mbnl1KO_Thymus_2 | Нокаут Mbnl1 | Представитель 2 | Тимус | Сопряженный конец | 100 бит/с | |
| GSM4116220 | СРР10261603 | Mbnl1KO_Thymus_3 | Нокаут Mbnl1 | Представитель 3 | Тимус | Сопряженный конец | 100 бит/с | |
| GSM4116221 | СРР10261604 | WT_Thymus_1 | Дикий тип | Представитель 1 | Тимус | Сопряженный конец | 100 бит/с | |
| GSM4116222 | СРР10261605 | WT_Thymus_2 | Дикий тип | Представитель 2 | Тимус | Сопряженный конец | 100 бит/с | |
| GSM4116223 | СРР10261606 | WT_Thymus_3 | Дикий тип | Представитель 3 | Тимус | Сопряженный конец | 100 бит/с | |
| 3П-Сек | GSM1480973 | СРР1553129 | WT_1 | Дикий тип (WT) | Представитель 1 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 40 бит/с |
| GSM1480974 | СРР1553130 | WT_2 | Дикий тип (WT) | Представитель 2 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 40 бит/с | |
| GSM1480975 | СРР1553131 | DKO_1 | Mbnl 1/2 двойной нокаут (DKO) | Представитель 1 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 40 бит/с | |
| GSM1480976 | СРР1553132 | DKO_2 | Mbnl 1/2 двойной нокаут (DKO) | Представитель 2 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 40 бит/с | |
| GSM1480977 | СРР1553133 | DKOsiRNA_1 | Mbnl 1/2 двойной нокаут с Mbnl 3 siRNA (KD) | Представитель 1 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 40 бит/с | |
| GSM1480978 | СРР1553134 | DKOsiRNA_2 | Mbnl 1/2 двойной нокаут с Mbnl 3 siRNA (KD) | Представитель 2 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 36 бит/с | |
| GSM1480979 | СРР1553135 | DKONTsiRNA_1 | Mbnl 1/2 двойной нокаут с нецелевым siRNA (Ctrl) | Представитель 1 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 40 бит/с | |
| GSM1480980 | СРР1553136 | DKONTsiRNA_2 | Mbnl 1/2 двойной нокаут с нецелевым siRNA (Ctrl) | Представитель 2 | Мышиные эмбриональные фибробласты (MEF) | Однокомнатный | 40 бит/с |
Таблица 1. Резюме наборов данных RNA-Seq и PolyA-seq, используемых для анализа.
