RNA-seq is in de loop der jaren op grote schaal gebruikt, meestal voor het schatten van differentiële genexpressie en genontdekking1. Bovendien kan het ook worden gebruikt om het gebruik op exonniveau te schatten als gevolg van gen dat verschillende isovormen tot expressie brengt, waardoor het bijdraagt aan een beter begrip van genregulatie op post-transcriptioneel niveau. De meerderheid van de eukaryote genen genereren verschillende isovormen door alternatieve splicing (AS) om de diversiteit van mRNA-expressie te vergroten. AS-gebeurtenissen kunnen worden onderverdeeld in verschillende patronen: het overslaan van complete exonen (SE) waarbij een (“cassette”) exon volledig uit het transcript wordt verwijderd samen met de flankerende introns; alternatieve (donor) 5′ splice site selection (A5SS) en alternatieve 3′ (acceptor) splice site selection (A3SS) wanneer twee of meer splice sites aanwezig zijn aan beide uiteinden van een exon; retentie van introns (RI) wanneer een intron wordt bewaard binnen het volwassen mRNA-transcript en wederzijdse uitsluiting van exongebruik (MXE) waarbij slechts één van de twee beschikbare exonen tegelijk kan worden bewaard 2,3. Alternatieve polyadenylering (APA) speelt ook een belangrijke rol bij het reguleren van genexpressie met behulp van alternatieve poly (A) -sites om meerdere mRNA-isovormen te genereren uit een enkel transcript4. De meeste polyadenylation sites (pa’s) bevinden zich in het 3′ onvertaalde gebied (3′ UTR’s), waardoor mRNA-isovormen met diverse 3′ UTR-lengtes worden gegenereerd. Aangezien de 3′ UTR de centrale hub is voor het herkennen van regulerende elementen, kunnen verschillende 3′ UTR-lengtes van invloed zijn op mRNA-lokalisatie, stabiliteit en translatie5. Er zijn een klasse van 3′-eindsequencingtests die zijn geoptimaliseerd om APA te detecteren die verschillen in de details van het protocol6. De hier beschreven pijplijn is ontworpen voor PolyA-seq, maar kan worden aangepast voor andere protocollen zoals beschreven.
In deze studie presenteren we een pijplijn van differentiële exon-analysemethoden 7,8 (figuur 1), die kunnen worden onderverdeeld in twee brede categorieën: exon-gebaseerd (DEXSeq9, diffSplice10) en gebeurtenisgebaseerd (multivariate analyse van transcriptsplitsing (rMATS)11). De op exon gebaseerde methoden vergelijken de vouwverandering over de omstandigheden van individuele exonen, met een maat voor de totale genplooiverandering om differentieel tot expressie gebracht exongebruik aan te roepen, en berekenen op basis daarvan een maat op genniveau van AS-activiteit. Op gebeurtenissen gebaseerde methoden gebruiken exon-intron-spanning junction reads om specifieke splicinggebeurtenissen zoals exon-overslaan of retentie van introns te detecteren en te classificeren, en onderscheiden deze AS-typen in de uitgang3. Deze methoden bieden dus complementaire weergaven voor een volledige analyse van AS 12,13. We selecteerden DEXSeq (gebaseerd op het DESeq214 DGE-pakket) en diffSplice (gebaseerd op het Limma10 DGE-pakket) voor de studie omdat ze tot de meest gebruikte pakketten voor differentiële splicinganalyse behoren. rMATS werd gekozen als een populaire methode voor event-based analyse. Een andere populaire op gebeurtenissen gebaseerde methode is MISO (Mixture of Isoforms)1. Voor APA passen we de exon-gebaseerde aanpak aan.
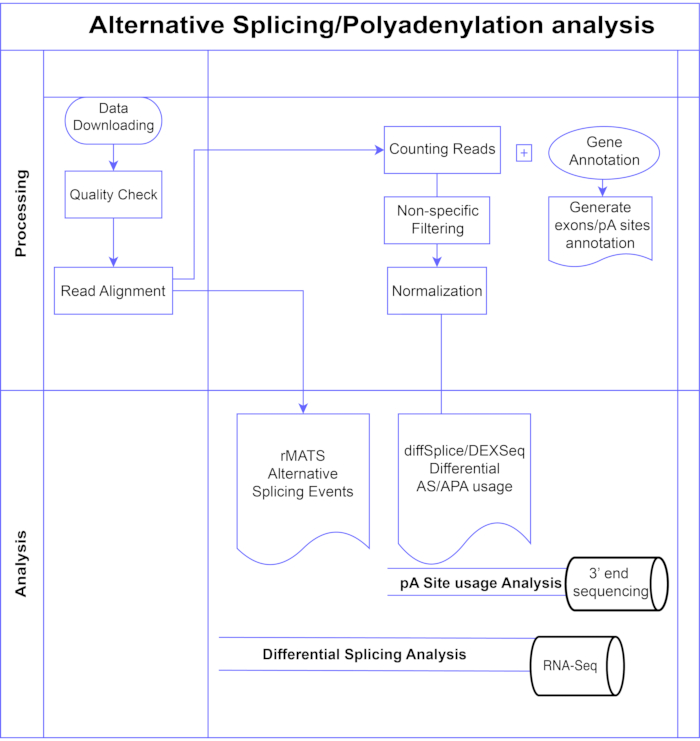
Figuur 1. Analysepijplijn. Stroomdiagram van de stappen die in de analyse zijn gebruikt. Stappen omvatten: het verkrijgen van de gegevens, het uitvoeren van kwaliteitscontroles en leesuitlijning gevolgd door het tellen van reads met behulp van annotaties voor bekende exonen, introns en pA-sites, filteren om lage tellingen en normalisatie te verwijderen. PolyA-seq-gegevens werden geanalyseerd voor alternatieve pA-locaties met behulp van diffSplice/DEXSeq-methoden, bulk RNA-Seq werd geanalyseerd voor alternatieve splicing op exon-niveau met diffSplice/DEXseq-methoden en AS-gebeurtenissen geanalyseerd met rMATS. Klik hier om een grotere versie van deze figuur te bekijken.
De RNA-seq-gegevens die in dit onderzoek zijn gebruikt, zijn verkregen uit Gene Expression Omnibus (GEO) (GSE138691)15. We gebruikten muis RNA-seq-gegevens uit deze studie met twee conditiegroepen: wild-type (WT) en Muscleblind-like type 1 knockout (Mbnl1 KO) met elk drie replicaties. Om de analyse van het gebruik van differentiële polyadenylation-sites aan te tonen, verkregen we polyA-seq-gegevens van muizenembryoblasten (MEF’s) (GEO Accession GSE60487)16. De gegevens hebben vier conditiegroepen: Wild-type (WT), Muscleblind-like type1/type 2 double knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO met Mbnl3 knockdown (KD) en Mbnl1/2 DKO met Mbnl3 control (Ctrl). Elke conditiegroep bestaat uit twee replicaties.
| GEO Toetreding | SRA Run nummer | Voorbeeldnaam | Conditie | Nabootsen | Weefsel | Sequencing | Leeslengte | |
| RNA-Seq | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 knock-out | Rep 1 | Thymus | Gekoppeld uiteinde | 100 bp |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 knock-out | Rep 2 | Thymus | Gekoppeld uiteinde | 100 bp | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 knock-out | Rep 3 | Thymus | Gekoppeld uiteinde | 100 bp | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | Wild type | Rep 1 | Thymus | Gekoppeld uiteinde | 100 bp | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | Wild type | Rep 2 | Thymus | Gekoppeld uiteinde | 100 bp | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | Wild type | Rep 3 | Thymus | Gekoppeld uiteinde | 100 bp | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | Wild type (WT) | Rep 1 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 40 bp |
| GSM1480974 | SRR1553130 | WT_2 | Wild type (WT) | Rep 2 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 40 bp | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 dubbele knock-out (DKO) | Rep 1 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 40 bp | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 dubbele knock-out (DKO) | Rep 2 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 40 bp | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 dubbele knock-out met Mbnl 3 siRNA (KD) | Rep 1 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 40 bp | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 dubbele knock-out met Mbnl 3 siRNA (KD) | Rep 2 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 36 bp | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 dubbele knock-out met non-targeting siRNA (Ctrl) | Rep 1 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 40 bp | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 dubbele knock-out met non-targeting siRNA (Ctrl) | Rep 2 | Embryonale fibroblasten bij muizen (MEF’s) | Single-end | 40 bp |
Tabel 1. Samenvatting van RNA-Seq en PolyA-seq datasets die voor de analyse zijn gebruikt.
