Este procedimento, que foi recentemente publicado no Nature Protocols1, foi citado mais de 3.000 vezes na literatura de arquivamento e tornou-se uma medida de rotina para caracterização de lignina e tanino, uma vez que fornece informações estruturais essenciais, rápidas e reprodutíveis.
Lignina e taninos
Quando a Química Verde foi introduzida por Paul T. Anastas e John C. Werner2,3, mudou drasticamente a concepção geral da Química. Em particular, destaca-se a importância de empregar materiais sustentáveis em vez de matérias-primas fósseis, como petróleo e carvão, como ponto departida,como ponto de partida, comopontodepartida. Entre os diferentes tipos de biomassa, a lignina é o biopolímero aromático mais abundante e pode ser vista como uma fonte potencial para commodities industriais e produtos de alto valor agregado4.
A lignina é a segunda mais abundante constituinte de madeira (com a celulose sendo a primeira e a hemicelulose em terceiro). Seu teor em plantas varia dependendo do tipo de planta: por exemplo, madeiras caracterizadas por uma menor quantidade de lignina em comparação com as madeiras macias (20% ± 4% vs. 28% ± 4%). Além disso, a distribuição de lignina dentro do tecido vegetal não é homogênea: o maior teor de lignina pode ser encontrado na parede celular5,6. A lignina é um material polifenólico obtido industrialmente como subproduto da indústria depapel/celulose 7. Recupera-se do processo de polpa de madeira, no qual as lascas de madeira são processadas principalmente na presença de CONDIÇÕES OH– e/ou OH– + HS– íon para separar a celulose da hemicellulose e da lignina (processos soda e/ou kraft)8,9.
As primeiras tentativas de estudo da lignina foram feitas por Payen e Schultze, respectivamente, em 1838 e 186510. Em 1977, Adler resumiu todo o conhecimento disponível da época11. Atualmente é reconhecido que os blocos de construção de lignina são três unidades fenil-propanoides: p-coumaryl, coniferyl e álcoois sinapyl. Esses monômeros, graças a um processo de polimerização radical livre, dão origem a unidades p-hidroxifenil, guaiacicl e sinapyl que eventualmente constituem amplamente a lignina(Figura 1)12. A falta de uma estrutura primária em ligninas implica uma dificuldade inerente à sua caracterização estrutural. Assim, a avaliação da distribuição do peso molecular sempre foi um tanto controversa. A lignina de madeira moída, a lignina isolada em condições leves que aproximam principalmente a protoligina10,é composta de oligômeros13 que interagem muito através dos processos de agregação supramolecular14,15.
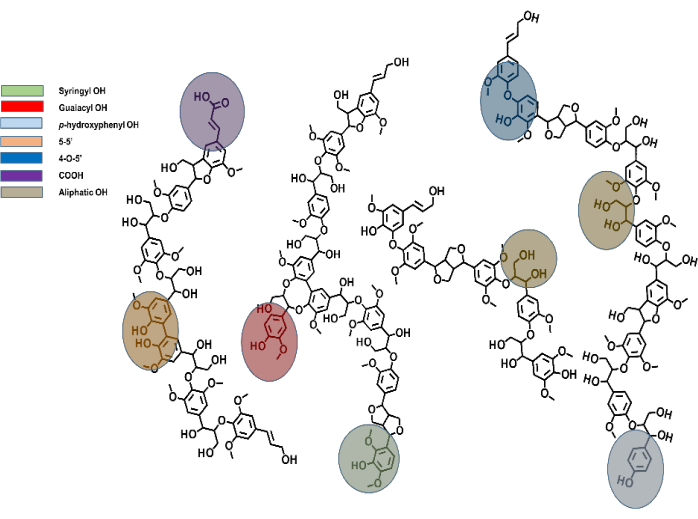
Figura 1: Um modelo representativo de lignina de madeira macia em que os diferentes tipos de títulos são destacados.
As ligninas são comumente classificadas dependendo de: (a) o tipo de madeira da qual são derivadas (por exemplo, madeira e madeira macia), (b) o processo usado para isolá-la. Os tipos de lignina industrial mais cruciais são Kraft, Lignosulfonates e Organosolv.
A estrutura da lignina é altamente dependente de sua origem e química de processamento. Mais especificamente, quando emerge a estrutura bastante complexa e irregular da lignina com sua diversidade natural e as complexas químicas de processamento, emerge um material de extrema variabilidade, diversidade e heterogeneidade, limitando seu uso a aplicações de baixo valor16. Enquanto as ligninas de madeira macia contêm principalmente unidades guaiacyl (G) com quantidades insignificantes de grupos de p-hidroxifenil (G lignin), as ligninas de madeira são compostas por subunidades guaiacyl e singyl (GS lignin) em diferentes proporções e ligninas de grama são constituídas por guaiacyl, subunidades de seringil e p-hidroxifenil(GSH lignin). A abordagem extrativista utilizada para o isolamento afeta drasticamente a estrutura da lignina emergente17. A Figura 2 retrata três estruturas de lignina, diferindo pela abordagem de isolamento empregada. Algumas considerações sobre o efeito do método de extração poderiam ser destacadas. Em primeiro lugar, a lignina kraft é uma lignina desfolada, altamente fragmentada e condensada, enquanto a lignina Organosolv tem uma estrutura semelhante à lignina de madeira moída (isolada usando a abordagem bjorkman)18,19,20. Por fim, os lignosulfonatos são caracterizados por um alto grau de sulfonação, dependendo da intensidade e das condições do processo de sulfonação extrativa.
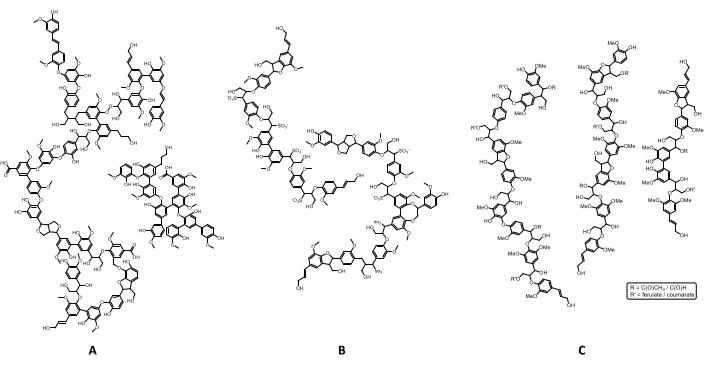
Figura 2: Estruturas representativas para ligninas técnicas. Neste número, podem ser observadas as diferenças entre os diferentes tipos de lignina. (A) A lignina kraft de madeira macia é altamente condensada, (B) lignosulfonatos são caracterizados por grupos sulfônicos em carbonos saturados, e(C) organosolv lignin tem uma estrutura semelhante à de lignina de madeira moída. Clique aqui para ver uma versão maior desta figura.
Semelhantes aos ligninas, os taninos são compostos polifenólicos que são encontrados nas plantas. Uma revisão recente e atualizada sobre as abordagens extrativítidas e aplicações dos taninos foi lançada recentemente por Das et al.21. A importância dos taninos no cotidiano pode ser destacada considerando dois exemplos: eles transmitem sabor e cor aos vinhos22; além disso, sua estrutura poli-fenólica oferece características antioxidantes e as torna ideais para aplicação na indústria de bronzeamento23. Os taninos são divididos em duas classes: hidrolavelmente e não hidrolízível. Taninos hidrolizantes podem ser considerados um polímero de ésteres ácidos gálicos, di-gálicos e ellagicos(Figura 3). Estes ésteres resultam da esterificação dos ácidos fenólicos com moléculas de açúcar (por exemplo, glicose, rhamnose e arabinose).
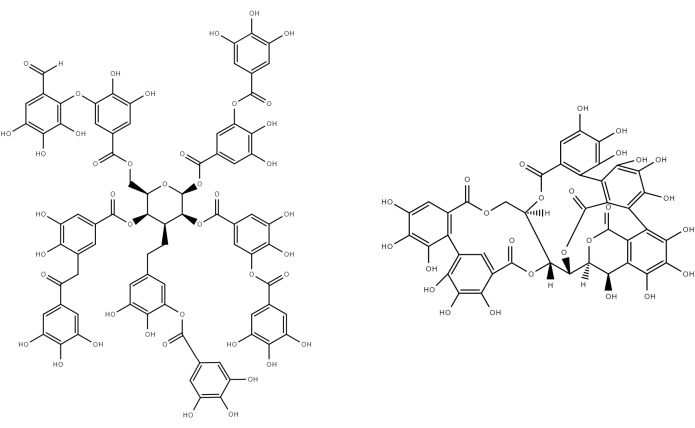
Figura 3: Taninos hidrolísegíveis típicos: ácido tânico, vescalgin. Por favor clique aqui para ver uma versão maior desta figura.
Taninos não hidrogavelmente, também conhecidos como taninos condensados, são polímeros e oligômeros derivados de flavan-3-ols. Entre flavan-3-ols, catequinas e gallocatechin são os mais frequentes. São compostos cristalinos incolores(Figura 4). A polimerização cria um polímero caracterizado por uma estrutura helicoidal. Os grupos hidroxis aromáticos são direcionados no exterior da hélice, enquanto os oxigênios piran estão no interior.
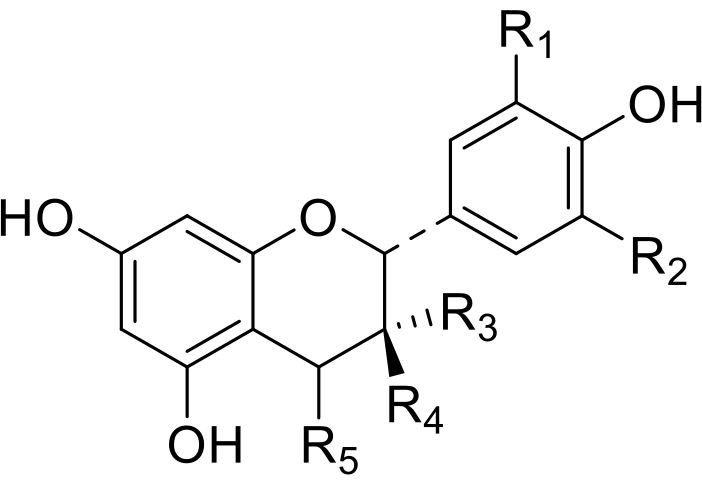
Figura 4: Estruturas de proantociaanatoidina: R =H, OH, OCH3. Clique aqui para ver uma versão maior desta figura.
Caracterização de ligninas e taninos usando RMN
Dois tipos de informação são cruciais na caracterização de lignina ou tanino: (a) estrutura química (por exemplo, teor de grupo hidroxy, natureza e frequência de ligações interunnitas) e (b) peso molecular e polidispersidade. Desde os primeiros estudos sobre lignina, diferentes técnicas têm sido empregadas para atingir esses objetivos, e duas classes de métodos surgiram: métodos químicos e físicos.
Na química da lignina, métodos químicos, como oxidação alcalina de nitrobenzeno, derivatização seguida de decote redutivo, oxidação permangásia e tiacidolise, têm sido historicamente amplamente utilizados24,25,26,27,28,29. No entanto, mesmo que os protocolos analíticos tenham sido implementados e otimizados, eles são exigentes, trabalhosos e exigem extensas habilidades experimentais30. Alternativamente, desde o início da análise instrumental, métodos físicos têm sido utilizados para realizar caracterizações de lignina e tanino31. Essas técnicas permitem superar os problemas dos métodos clássicos facilitando a caracterização da estrutura da lignina.
A Ressonância Magnética Nuclear (RMN) permite obter informações sobre estrutura de lignina e composição química entre as técnicas instrumentais. Em particular, os dados de espectros quantitativos monodimensionais 1H NMR e quantitativos 13C NMR podem fornecer informações sobre diferentes tipos de unicções interunnitasligina 32,33,34,35. Infelizmente, os espectros monodimensionais sofrem de sobreposição de sinal, o que pode prejudicar seriamente os esforços de integração de sinais. Versões quantitativas de HSQC (Coerência Quântica Única Heteronuclear), Q-HSQC (Quantitative – Heteronuclear Single Quantum Coherence), têm sido usadas para entender melhor a estrutura da lignina, fornecendo informações úteis sobre ligações internas. No entanto, eles não podem ser totalmente utilizados para determinar as várias unidades de edifícios13,36,37 quantitativamente.
Para superar as questões associadas à RMN mono e bidimensional, considerou-se a derivatização do substrato. Entre as vantagens dessa abordagem está que rótulos específicos podem ser introduzidos dentro da complexa macromolécula e nenhuma interferência espectral resulta do solvente no qual os substratos rotulados são dissolvidos1. Verkade foi a pioneira neste campo, realizando 31P NMR análise de derivados fosforosos, derivados de carvão e compostos relacionados38. Em sua publicação, foi realizada uma triagem de diferentes reagentes contendo fósforo (fósforo) e registro da mudança química de outros compostos rotulados. A equipe de Argyropoulos introduziu pela primeira vez a derivatização para a análise quantitativa e qualitativa de grupos hidroxy em lignina em 1991. Após estudar a derivatização de compostos modelo de lignina utilizando reagentes contendo fósforo, seu grupo abriu caminho para uma das técnicas mais utilizadas diariamente em química de lignina, 31P NMR análise39,40,41,42,43. Entre as diferentes fosfolanas examinadas, Argyropoulos chegou ao uso de 2-cloro-4,4,5,5-tetrametila-1,3-2-dioxaphospholane (TMDP) como sendo o mais adequado para realizar a análise de lignina44. O TMDP reage seletivamente com grupos hidroxis causando a formação quantitativa de derivados contendo fósforo caracterizados por mudanças químicas específicas de 31P NMR(Figura 5).
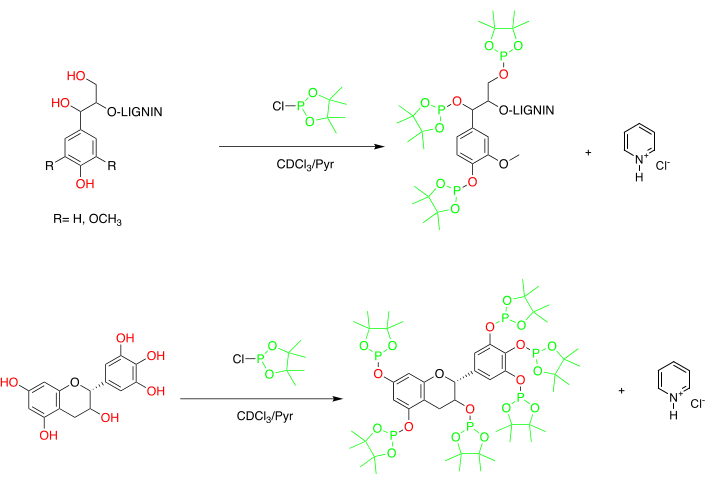
Figura 5: Química de fosfitilação de lignina e tanino. A rotulagem de grupos H de lignina e tanino labile É realizada pela reação in situ. Os polifenóis rotulados são caracterizados por bandas específicas de 31P NMR correspondentes aos diferentes tipos de grupos hidroxi. Clique aqui para ver uma versão maior desta figura.
A derivatização da amostra é realizada em uma mistura piridina/clorofórmio (1.6:1); esta escolha resulta de uma avaliação precisa. Piridina tem duas vantagens. Em primeiro lugar, selecionar um solvente caracterizado por um parâmetro Hildebrand de cerca de 22,1 MPa1/2 simplifica e amplifica a solubilização da lignina45. Consequentemente, a adição de piridina como solvente, cujo parâmetro Hildebrand equivale a 21,7, é, portanto, ótima. Em segundo lugar, a reação do TMDP com grupos hidroxi é acompanhada pela formação de ácido clorídrico (HCl) como subproduto com implicações negativas concomitantes para a formação fácil de derivados lignina-fosfolane. Por essa razão, o HCL resultante precisa ser neutralizado. Quando presente em excesso significativo, a basicidade da piridina, em relação ao TMDP, permite a neutralização do HCl (através da formação de cloridrato de piridina).
O uso do sistema de solvente binário de cloroforme recomendado/deuterado baseia-se em três razões. Em primeiro lugar, favorece a dissolução da amostra. Em segundo lugar, como o cloridrato de piridina é solúvel em clorofórmio, pode evitar a precipitação e a deterioração do espectro final. Em terceiro lugar, o clorofórmio deuterado é escolhido por seu sinal único único, permitindo o bloqueio do espectrômetro NMR durante o processo de aquisição. A derivatização da amostra é realizada na presença de um padrão interno. Dessa forma, quando a amostra e o padrão são derivatizados, a comparação das integrais dos picos da amostra e do padrão permite a quantificação da quantidade para cada tipo de grupo hidroxy presente. Vários compostos foram considerados como padrões internos. Esses compostos são caracterizados por um único grupo hidroxi por molécula, oferecendo um único sinal acentuado no espectro de 31P NMR após a derivatização. A seleção do padrão deve ser feita com cuidado. Seu sinal não deve se sobrepor aos da amostra derivatizada. O colesterol foi amplamente utilizado durante os primeiros dias. No entanto, uma sobreposição parcial com sinais decorrentes do grupo hidroxy alifático limita seu uso. Para análise de rotina, são preferidas soluções padrão internas de N-hidroxi-5-norbornene-2,3-dicarboximida (NHND). No entanto, devido à instabilidade do NHND, suas soluções padrão só podem ser armazenadas por alguns dias46.
O método descrito representa a implementação e otimização do protocolo analítico voltado à caracterização qualitativa e quantitativa de ligninas desenvolvida por Argyropoulos37,38,39,40,41,42. Comparado com muitas outras técnicas disponíveis para elucidação estrutural de lignina, o método tem sido amplamente aceito como sendo um dos mais fáceis, rápidos e reprodutíveis. A validade dos métodos químicos molhados (por exemplo, nitrobenzeno, oxidações permangês, etc.) baseia-se nas boas habilidades experimentais do operador, confinando efetivamente o método a operadores limitados. Além disso, não é incomum encontrar fatores de correção na literatura para métodos químicos molhados para explicar várias desvantagens. O protocolo NMR descrito de 31P não requer habilidades experimentais avançadas tornando este facilmente aplicável, fácil de usar e amplamente disponível. Em comparação com outros métodos analíticos instrumentais, 31P NMR é a única técnica capaz de detectar e quantificar precisamente os diferentes grupos hidroxi em ligninas. Por exemplo, ftir pode ser usado para identificar vários grupos hidroxi, como 1H NMR. Ambas as técnicas, no entanto, sofrem, pois não podem oferecer dados quantitativos confiáveis devido a extensos problemas de sobreposição de sinal. Outra técnica amplamente utilizada é a espectroscopia UV-Vis, relatada em primeiro lugar por Goldschmid. A abordagem, no entanto, limita-se a uma determinação geral geral dos grupos hidroxi, uma vez que não pode efetivamente diferenciar-se entre os OHs alifáticos, aromáticos e carboxílicos47.
Do ponto de vista econômico, a única limitação da técnica de 31P NMR é o preço do TMDP, que é um reagente relativamente caro. Custa cerca de 190 USD por grama; consequentemente, se o custo de análise fosse aproximado apenas ao preço do TMDP, excluindo aqueles provenientes da mistura piridina/clorofórmio e os do tempo do operador, seria de cerca de 24 USD por análise. Para resolver esse problema, muitos laboratórios recorrem à sintetização do TMDP, reduzindo assim os custos do reagente. Para isso, pinacol e tricloreto de fósforo são reagidos na presença de trietilamina44. Tecnicamente, essa reação é relativamente fácil; no entanto, é necessário cuidado com o uso de triclorito fosforoso e seu trabalho, incluindo destilação a vácuo bem controlada. Mais detalhes sobre a síntese do TMDP podem ser fornecidos mediante solicitação.
Embora este protocolo esteja entre os melhores em termos de facilidade, reprodutibilidade e precisão, alguns pontos críticos precisam ser destacados. Em primeiro lugar, a amostra precisa ser totalmente solúvel na mistura piridina/clorofórmio identificada. Essa consideração é fundamental porque a reação quantitativa de fosfiscimento dos grupos hidroxil precisa ocorrer em condições completamente homogêneas. Se apenas parte da amostra for solubilizada, a análise resultante seria imprecisa. Em segundo lugar, a amostra a ser examinada precisa ser livre de umidade e solventes, uma vez que essas variáveis afetarão prejudicialmente a precisão e o sucesso geral da análise. Traços de umidade reagirão com o TMDP dando 2-hidroxi-4,4′-5,5′-tetramethyl-1,3,2-dioxaphospholane. Este composto é um sal floculante amarelo-pálido, insolúvel na mistura piridina/solvente de clorofórmio, causando aquisição inadequada de sinal NMR. Uma vez que apenas um pequeno peso (~30 mg) de uma amostra é necessário, ele precisa estar livre de voláteis para que seu peso preciso seja conhecido com precisão antes da análise.
Às vezes, problemas de solvação amostral podem ser promovidos (especialmente para amostras altamente oxidadas) adicionando pequenas quantidades de um co-solvente (ou seja, dimetilformamida), auxiliando a dissolução da amostra. Em princípio, todos os solventes que não interagem com o TMDP podem ser usados para ajudar a dissolução da amostra. A eleição de um co-solvente não pode incluir co-solventes contendo hidroxi labile ou grupos de amino, uma vez que eles reagem com o reagente, causando espectros finais enganosos. Notavelmente, dimetilsulfoxida também reage com o TMDP impedindo seu uso como co-solvente. Líquidos iônicos à base de piridina, como cloreto de 1-alil-3-butylpyridinium, podem ser usados quando surgirem problemas de solubilidade; no entanto, o líquido iônico deve ser novamente seco48. Para dissolver lignosulfonatos (um tipo de lignina caracterizado por um alto grau de sulfonação), demonstrou-se um pré-tratamento envolvendo a conversão de grupos neutralizados em sua forma ácida. Lignosulfonates podem ser convenientemente convertidos em suas condições ácidas usando resinas de troca ácidas em mídia aquosa. Os ácidos lignosulfônicos resultantes são isolados da solução por sua adsorção em resinas específicas (por exemplo, XAD-7) e desorção no etanol. A evaporação das soluções etanolicas sobre a pressão reduzida a 40 °C permite o isolamento dos ácidos lignosulfônicos. Essas ligninas podem então ser caracterizadas por 31P NMR porque são solúveis na mistura piridina/clorofórmio proposta pelo protocolo.
A secagem prolongada de vácuo a temperaturas amenas reduz efetivamente a quantidade de umidade e outros voláteis em cada amostra. Notavelmente, pequenas quantidades de água não afetam o espectro final porque o TMDP é adicionado em excesso. Além disso, em alguns casos, uma pequena quantidade de 2-hidroxi-4,4′-5,5′-tetramethyl-1,3,2-dioxaphospholane pode resultar da umidade presente no tubo NMR ou no frasco amostral. Nestes casos, a agitação é suficiente para dissolver completamente a quantidade do precipitado formado. Se for formada uma alta quantidade de 2-hidroxi-4,4′-5,5′-tetrametil-1,3,2-dioxaphospholane, sugere-se que repita a preparação da amostra, melhorando o tratamento de secagem. Por exemplo, antes de usar, todos os vidros podem ser brevemente aquecidos com uma arma de calor.
A faixa espectral utilizada para registrar o espectro é ampla em comparação com a região de interesse para o sinal em relação aos diferentes grupos hidroxil. No entanto, isso é obrigatório para entender se a derivatização da amostra ocorreu com sucesso. A confirmação da derivatização completa da amostra é dada pela presença de um sinal forte em torno de 174 ppm. Este pico acentuado deve-se ao TMDP não redigido, e sua existência garante que o reagente estava presente em excesso e, portanto, todos os grupos de hidroxil foram derivados. Se este pico estiver ausente, as duas causas mais prováveis são: (1) a quantidade de TMDP utilizada é insuficiente para realizar a derivatização completa da amostra, ou (2) uma alta quantidade de água está presente na amostra. No primeiro caso, o uso de uma quantidade maior de TMDP provavelmente garantiria a derivatização completa da amostra, e o sinal a 174 ppm apareceria. No segundo caso, a amostra deve ser seca mais extensivamente. Uma vez que um excesso de TMDP é assegurado, a integração de pico pode ser realizada. Antes desta operação, zoom para uma janela mais estreita (150 a 132 ppm) que limita os sinais de interesse.
A quantidade de amostra (~30 mgs) a ser analisada, relatada no protocolo experimental acima, foi selecionada para coletar espectrômetros de boa qualidade para um espectrômetro NMR de 300 MHz ou mais. No entanto, observamos que é possível reduzir a quantidade amostral se um ímã de campo de 500 MHz ou mais alto for usado. Por exemplo, na Figura 8D, o espectro NMR (resultante de um instrumento de 700 MHz) de uma amostra preparada com 7,2 mg de lignina é mostrado. A integração de sinais deste espectro oferece os mesmos resultados obtidos ao utilizar maiores quantidades de lignina. Este fato amplifica a aplicabilidade deste protocolo para todas as pesquisas em que pequenas quantidades de produtos estão disponíveis.
No geral, este protocolo experimental pode ser aplicado a muitas aplicações de pesquisa e desenvolvimento ao entender a origem e o destino dos vários grupos hidroxy presentes em ligninas e taninos. Em particular, quando juntamente com os dados GPC e HSQC, os dados resultantes oferecem a oportunidade de elaborar e especular mais sobre a estrutura da lignina ou um tanino. Em muitos casos em que modificações químicas são aplicadas aos grupos hidroxidos de lignina ou um tanino, análises quantitativas de 31P NMR podem ser extremamente valiosas para detectar se essas modificações ocorreram e em que grau. Por exemplo, a Figura 9 mostra dois espectros de RMR da mesma lignina antes e depois de sua oxidação. Uma avaliação qualitativa simples mostra a redução de grupos hidroxis alifáticos e aromáticos na oxidação, fornecendo informações e orientações valiosas.
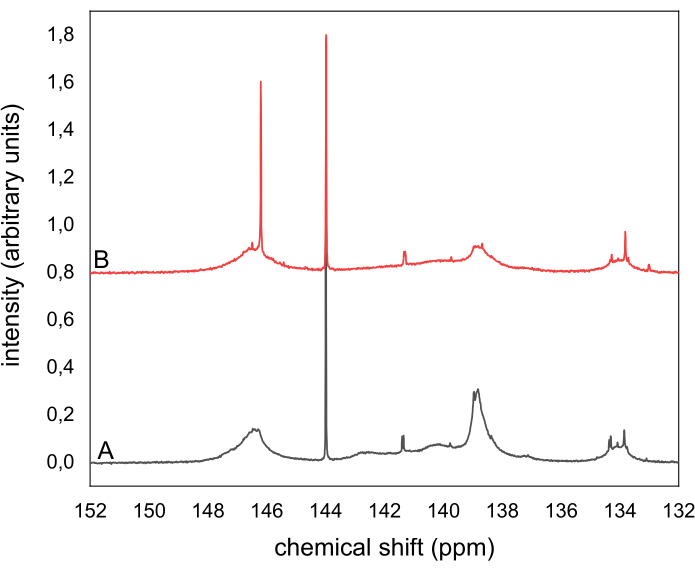
Figura 9: Espectros quantitativos de 31P NMR do mesmo Organosolv liginogino derivado utilizando TMDP (A) Anterior e (B) postam sua oxidação. Os espectros foram registrados utilizando um espectrômetro de 300 NMR. Clique aqui para ver uma versão maior desta figura.
Em conclusão, essa técnica tem todos os atributos de estar entre as ferramentas mais essenciais e poderosas quando as consultas que lidam com ligninas polifenólicas, OH com ligninas e taninos (e até polímeros sintéticos)49,50,51 precisam ser feitas em uma variedade de campos, que vão da química à engenharia, da biologia ao polímero, e aplicações farmacêuticas.
