This protocol contains the detailed steps for generating homozygous mutants of the migratory locusts with the RNP consisting of Cas9 protein and in vitro synthesized sgRNA. The following are some representative results of CRISPR/Cas9-mediated target gene knockout in locusts, including target selection, sgRNA synthesis and verification (Figure 1A), egg collection and injection, mutant screening and passaging, cryopreservation, and resuscitation of the homozygous eggs.
In this study, the target site for CRISPR/Cas9 system is selected according to the results of three online programs (the E-CRISP, CRISPOR, and ZiFit) and located in the first exon (Figure 1B). According to the Cas9 cleavage assay in vitro (Table 1), the CRISPR/Cas9 RNP could digest the PCR fragment (containing the target site) with a cleavage rate of about 55% (Figure 1C). Then, this RNP was microinjected into 120 fertilized eggs at their single-cell stage using the standard microinjection system (Figure 2 and Figure 3). The sequencing results for embryonic stage mutation rate estimation suggested efficient genome editing at the target site (Figure 4A). Further, this study resulted in a 52.73% nymph hatching rate and 66.7% of the G0 adults were mosaic mutants (Table 4).
Further, the G0 chimeras were crossed with wild-type locusts to obtain heterozygous G1 individuals and the G1 heterozygotes with the same mutations (screened by TA-cloning) were in-crossed to generate the G2 animals. PCR-based phenotyping was used for detecting mutants and the obtained homozygotes were in-crossed to establish stable mutant lines (Figure 4B).
Meanwhile, the excess homozygous eggs were cryopreserved to improve the utilization rate of the homozygotes. Although the hatching rate of the cryopreserved eggs was reduced with the extension of cryopreservation time (Table 5), the remedial actions including applying filter paper fragments for keeping eggs wet and recovering these preserved eggs with a gradient of rising temperature were both helpful for the resuscitation (Figure 5 and Table 5). Finally, the homozygous population of mutants was successfully kept for subsequent research.
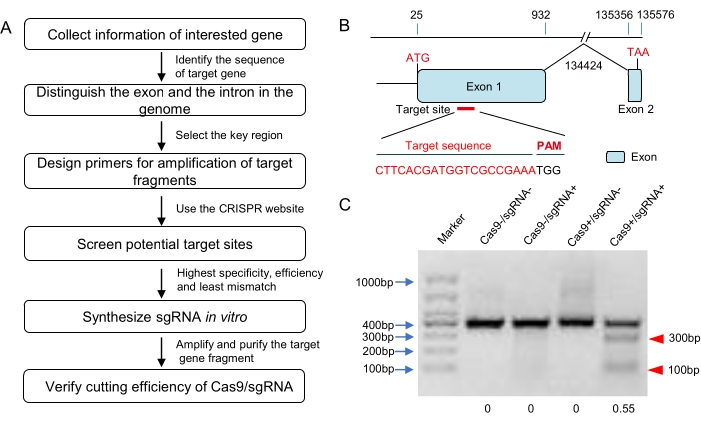
Figure 1: Design of the target and in vitro verification of the sgRNA. (A) The flow diagram for target selection, sgRNA synthesis, and bioactivity verification in vitro (including but not limited to locusts). (B) Schematic diagram of the gene structure and target site. The exons of this target gene are shown as blue domains and the target site was selected downstream of the start codon in exon 1. The target sequence is highlighted in red. (C) The agarose gel electrophoresis result of in vitro Cas9 cleavage assay. A DNA fragment harboring the target sequence (about 400 bp in length) was amplified and used as the substrate for Cas9 digestion. The expected small bands (about 100 bp and 300 bp here; marked with red triangles) suggested that the synthesized sgRNA could induce effective Cas9 cleavage. The cleavage rate was about 55% according to the grayscale analysis. Please click here to view a larger version of this figure.
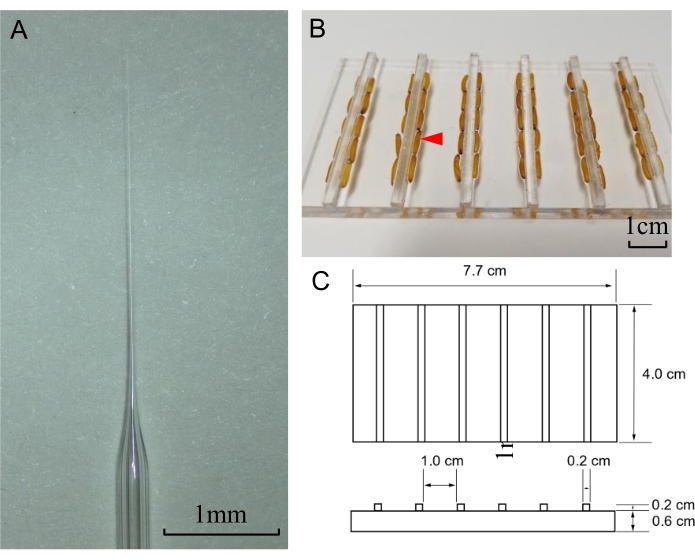
Figure 2: Needle preparation and the microinjection pad for locust eggs. (A) Tip of a prepared needle for microinjection of locust eggs. Scale bar: 1 mm. (B) Picture of a microinjection pad with eggs (indicated with a red triangle) arranged on it. The scale bar represents 1 cm. (C) The size of the microinjection pad. Please click here to view a larger version of this figure.

Figure 3: Egg microinjections. (A) A representative microinjection system labeled with red boxes indicating a microscope (middle) and a micromanipulator (right) connected to a microinjector (left). The computer is used for data storage and for providing auxiliary observation. (B) Injection of the locust egg. The injection site is marked with a red triangle. (C) A hatched nymph under the microscope. The scale bars represent 1 mm. Please click here to view a larger version of this figure.
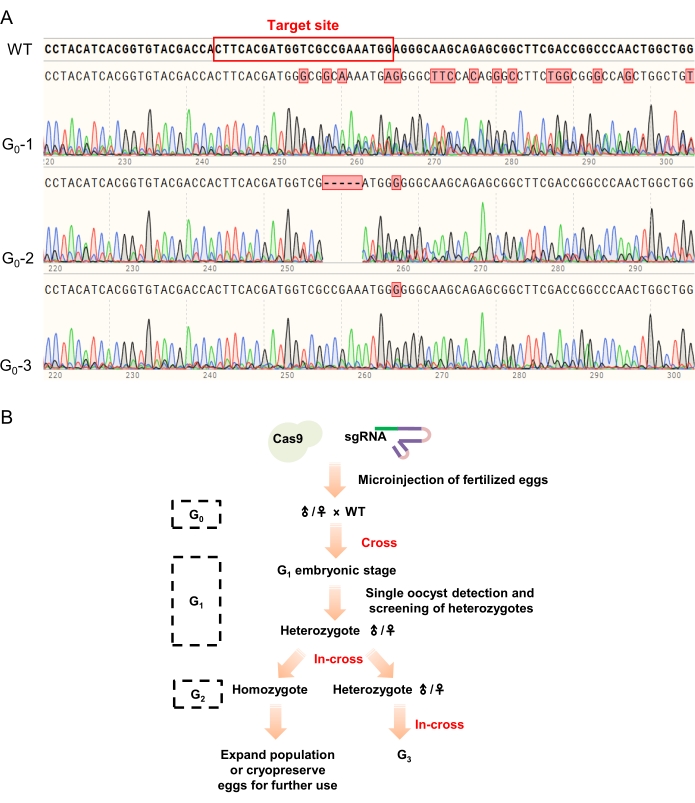
Figure 4: The sequencing results of G0 animals and passaging strategy of the mutant lines. (A) The typical sequencing results in the G0 screening. Multiple peaks near the target site (marked with a red rectangle in the wild-type sequence) indicated that the tested egg/locust was successfully edited by CRISPR/Cas9 system (as shown in G0-1 and G0-2), while the individual without any change in the sequencing result was considered as not edited and abandoned (e.g., G0-3). (B) The passaging strategy of the mutant lines. The G0 animals were firstly screened by PCR-mediated genotyping and those with multiple peaks in their sequencing results were crossed with wild-type locusts to generate the G1 animals. PCR-mediated genotyping and TA cloning were used to identify the G1 heterozygotes. Then, heterozygotes with the same mutations were in-crossed to generate the G2 animals (homozygotes and heterozygotes) for further research and passaging. Please click here to view a larger version of this figure.
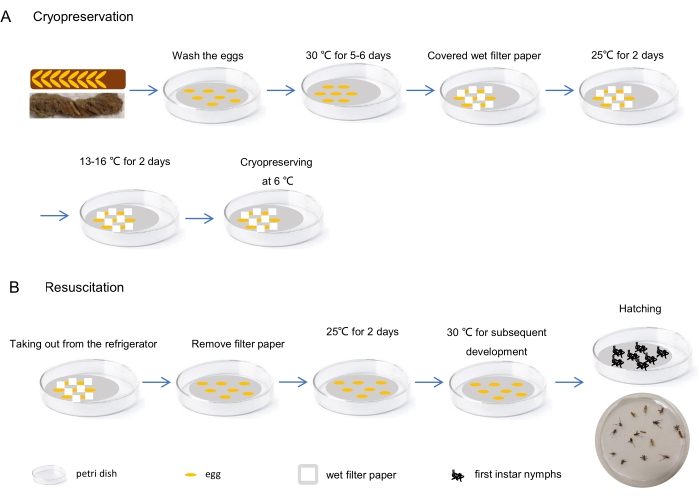
Figure 5: Egg cryopreservation and resuscitation. (A) The procedure of cryopreservation: Isolate the eggs from egg pods and incubate them at 30 °C for 5-6 days after washing with sterile water. Then, gather the developed eggs together in the Petri dish and cover them with small scraps of moist filter paper. Wrap the entire Petri dish with a paraffin film and keep it at 25 °C for 2 days, followed by another 2 days at a relatively lower temperature (e.g., 13-16 °C). Finally, refrigerate it at 6 °C. (B) The procedure of resuscitation: Take the Petri dishes with cryopreserved eggs out from the refrigerator and keep them at 25 °C for 2 days after removing the filter paper scraps. Then, the eggs can be cultured for subsequent development at 30 °C until the nymphs hatch. Please click here to view a larger version of this figure.
| Reagent | Volume (μL) |
| Cas9 protein (300 ng/μL) | 1 |
| sgRNA (300 ng/μL) | 1 |
| PCR product (200 ng/μL) | 1 |
| 10×NEBuffer r3.1 | 1 |
| Nuclease-free water | 6 |
Table 1: In vitro digestion system. 200 ng of purified target fragment (PCR product) was mixed with the CRISPR/Cas9 system (300 ng of each component) for testing the bio-activity of the synthesized sgRNAs. A commercial buffer was used and nuclease-free water was added to make up the total volume to 10 µL.
| Reagent | Volume (μL) |
| 2xEs Taq MasterMix | 12.5 |
| Forward primer | 0.5 |
| Reverse primer | 0.5 |
| Template (lysis product) | 1 |
| Nuclease-free water | 10.5 |
Table 2: PCR system for target gene fragment amplification. To amplify the genomic fragment of the target gene, a commercial Taq mix was used and primers were added according to the manufacturer's instructions. 1 µL of the lysis product was used as the template and nuclease-free water was used to make the total volume to 25 µL.
| Temperature (°C) | Time | Cycles |
| 95 | 5 min | 1 |
| 95 | 30 s | 35 |
| 55 | 30 s | |
| 72 | 40 s | |
| 72 | 10 min | 1 |
Table 3: PCR program for target gene amplification. The PCR program for target gene amplification was: 95 °C for 5 min; 35 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 40 s; 72 °C for 10 min.
| Items | Data |
| No. of injected eggs | 120 |
| Tested embryos | 10 |
| No. of hatched nymphs | 58 |
| Hatching rate | 52.73% |
| No. of G0 adult | 12 |
| No. of G0 mutants | 8 |
| Mutation efficiency in G0 adults | 66.67% |
Table 4: Summary of editing efficiency. 120 eggs were injected to knock out the target gene and 10 eggs were taken for testing during the embryo stage. 58 nymphs hatched from the rest embryos (the hatching rate was 52.73%). Finally, 12 G0 locusts developed to the adult stage and 8 of them were successfully edited at the target site. The mutation rate in G0 adults was 66.67%.
| Control group | Group 1 | Group 2 | Group 3 | Group 4 | |
| No. of eggs | 120 | 120 | 120 | 120 | 120 |
| Temperature treatment for storage | 30 °C | 30°C (5-6 d)→6 °C | 30°C (5-6 d)→25°C (2 d)→13-16°C (2 d)→6 °C | 30°C (5-6 d)→25°C (2 d)→13-16°C (2 d)→6 °C | 30°C (5-6 d)→25°C (2 d)→13-16°C (2 d)→6 °C |
| Storage time | 14 days | 14 days | 1 month | 3 months | 5 months |
| Temperature treatment for resuscitation | 30 °C | 6°C→30 °C | 6°C→25°C (2 d)→30 °C | 6°C→25°C (2 d)→30 °C | 6°C→25°C (2 d)→30 °C |
| No. of hatched locusts | 108 | 0 | 96 | 86 | 78 |
| Hatching rate | 90.00% | 0 | 80.00% | 71.67% | 65.00% |
| No. of adult locusts | 81 | 0 | 60 | 46 | 31 |
| Eclosion rate | 75.00% | 0 | 62.50% | 53.49% | 39.74% |
Table 5: Summary of the cryopreservation and resuscitation of eggs. 600 eggs were divided into five groups for cryopreservation and resuscitation study. The first group (control) of 120 eggs was always incubated at 30 °C and stored for 14 days. The second group (Group 1) of 120 eggs was transferred to a refrigerator after incubation at 30 °C for 5-6 days and stored at 6 °C for 14 days. Then, these eggs were transferred to 30 °C for resuscitation. The other groups (group 2, group 3, and group 4, each group contained 120 eggs) experienced a gradient cooling and recovering temperature treatment as described in the protocol (steps 6.1-6.3) with a different storage time (1 month for group 2, 3 months for group 3, and 5 months for group 4). At last, 108 nymphs hatched from the control group (at a hatching rate of 90%) and 81 of them developed to the adult stage (75% of the eclosion rate). No locusts hatched in the second group (Group 1). The hatching rate and eclosion rate of the other groups were lower compared to the control group and declined with the storage time. 96 nymphs hatched in group 2 and 60 of them developed to the adult stage. 86 nymphs hatched in group 3 and 46 of them developed to the adult stage. In group 4, 78 nymphs hatched and 31 of them developed to the adult stage.
