La microscopía SRS de banda ancha es una poderosa técnica de imagen que ofrece un auténtico contraste químico para identificar y desenredar los componentes químicos de una muestra heterogénea. El potencial de esta herramienta analítica puede ser beneficioso para varios campos de investigación, que van desde la ciencia de los materiales hasta la histopatología. La desventaja de la microscopía SRS de banda ancha es el hecho de que es técnicamente exigente; el experimentalista no solo requiere conocimientos sobre fuentes láser de banda ancha, sino que también necesita manipular los pulsos láser para generar SRS de manera eficiente, una señal que, a su vez, debe medirse con sofisticados esquemas de detección. Este documento presenta un protocolo que describe un flujo de trabajo para producir mapas químicos de compuestos químicos mixtos utilizando un microscopio SRS de banda ancha multiplex. Aunque el trabajo descrito puede ser trivial para algunos físicos láser y microscopistas no lineales, puede que no sea el caso para los lectores interesados en los beneficios de la microscopía SRS de banda ancha cuyo conocimiento científico reside fuera de estos dominios. Por lo tanto, nuestro objetivo fue detallar cada paso para guiar a la amplia audiencia interesada en la microscopía SRS de banda ancha.
El protocolo en cuestión comenzó mostrando cómo preparar una muestra simple pero espectroscópicamente rica compuesta de varios dispersores Raman fuertes y conocidos. Discutimos cómo obtener la bomba de banda ancha y los haces Stokes de banda estrecha necesarios para configurar un microscopio SRS. La Figura 5C muestra un esquema de las configuraciones SHG y OPO. Tenga en cuenta que la lente f1 enfoca el haz fundamental en LBO1 para generar el SHG, mientras que un espejo dicroico refleja la radiación SHG y transmite el haz fundamental residual. Una segunda lente f2 colima el haz SHG. Como f2 > f1, el haz SHG se expande por un factor igual a f2/f1. Una tercera lente f3 enfoca el haz SHG expandido en un segundo cristal LBO Tipo I (LBO2) cortado a θ = 90° y φ = 29.0°. Al bombear LBO2 con el mencionado SGH (520 nm), la radiación dentro del rango de 680-910 nm surgirá de LBO2 a través de la generación de diferencia de frecuencia (DFG), produciendo dos haces: la señal y el ralentí27 (Figura 5D, E). Este último se descarta, mientras que el primero se amplifica en la cavidad OPO para entregar los pulsos de bomba empleados en los experimentos SRS. La bomba del OPO a 520 nm, es decir, el haz SHG, no debe confundirse con la bomba de los experimentos SRS (es decir, el haz de señal del OPO).
El contraste en la microscopía SRS se origina a partir de una señal no lineal generada en el punto focal del microscopio, una señal que exige confinar un gran número de fotones en el plano de la muestra en un momento dado. Este confinamiento de fotones se logra con un objetivo de microscopio de alta apertura numérica (NA), una matriz de lentes que también establece la resolución espacial del sistema: cuanto mayor es el NA, mayor es la resolución espacial. Sin embargo, los objetivos de NA altos están densamente llenos de vidrio, que introduce GDD positivo a la radiación pulsada, un chirrido de frecuencia que finalmente amplía el perfil temporal de los pulsos39. Por lo tanto, el GDD introducido por el objetivo del microscopio podría aumentar la duración de los pulsos de la bomba de banda ancha, haciéndolo aún más largo que la envolvente temporal de Stokes y reduciendo el ancho de banda efectivo y accesible de la señal Raman. Además, esta ampliación también podría introducir una distorsión del perfil espectral del espectro SRS medido.
En CARS, la señal espectroscópicamente relevante emerge en longitudes de onda que difieren de las de los campos de excitación. Se puede utilizar un simple tubo fotomultiplicador o una cámara de dispositivo de carga acoplada (CCD) para integrar la señal CARS en el tiempo, resumiendo miles de pulsos para promediar el ruido del láser. En cambio, la señal SRS aparece como una débil transferencia de modulación incrustada dentro de un fondo láser fuerte y fluctuante. Debido a que esta modulación es débil, el ruido del láser puede abrumarlo fácilmente, reduciendo tanto la velocidad de imagen como la sensibilidad del microscopio SRS. Por lo tanto, antes de obtener imágenes, es imperativo medir el ruido de intensidad relativa (RIN) para determinar si el láser es adecuado para imágenes SRS de alta velocidad y seleccionar la frecuencia de modulación con el ruido más bajo. El RIN se define como la densidad espectral de potencia de ruido [δP(f), con unidades W2/Hz] del láser normalizada por la potencia óptica media (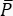 )40,41. En otras palabras, el RIN describe las fluctuaciones normalizadas del láser a diferentes frecuencias (Eq [4]).
)40,41. En otras palabras, el RIN describe las fluctuaciones normalizadas del láser a diferentes frecuencias (Eq [4]).
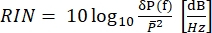 (4)
(4)
Por lo tanto, el RIN es un parámetro del sistema SRS que determina el rango de frecuencia de modulación ideal para los experimentos. Por ejemplo, la barra de olivo en la Figura 8 muestra el rango de frecuencia de modulación ideal para imágenes SRS. En el caso de SRS de banda estrecha, el usuario debe medir el RIN tanto de la bomba como de Stokes para elegir qué haz necesita ser modulado para lograr un rendimiento óptimo. Tenga en cuenta de la Figura 8, por ejemplo, que el haz de Stokes tiene un RIN ligeramente más alto que la bomba, lo que implica que las mediciones de SRG resultarían más ruidosas que sus contrapartes SRL. En el caso del SRS de banda ancha, el haz que debe modularse es el haz de banda estrecha.
La dispersión angular D de la rejilla expresa el ángulo de difracción en función de la longitud de onda, y se define como la derivada de la ecuación de rejilla. Para la configuración littrow, la dispersión angular viene dada por Eq (5).
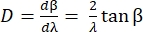 (5)
(5)
Para obtener Eq (5), asumimos α = β, resolvimos Eq (2) para m/d e insertamos el resultado en dβ/ dλ. Nótese que en la configuración de Littrow, β = sin-1(mλ/2d). Dentro de la aproximación de ángulo pequeño, el cambio de posición a lo largo del espectro es fdβ ≈ dl (Figura 10). Así, insertando dβ en Eq (5), podemos calcular la dispersión lineal, una cantidad con unidades de nm mm-1 usando Eq (6):
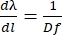 (6)
(6)
Para una rejilla de difracción que funciona en la configuración Littrow con 1.851,85 ranuras/mm, d = 540 nm. Si usamos la difracción de primer orden de la luz a ~789 nm, D = 0.0027 rad nm-1. Con una lente f = 750 mm, obtenemos una dispersión lineal de ≈ 0,5 nm mm-1, lo que se traduce en ≈ 7,8 cm-1 mm-1. Así, la distancia focal de la lente determina la “densidad” de nm por mm en el plano del detector: cuanto mayor es la distancia focal, menos nm por mm se obtiene, aumentando el espacio entre las líneas espectrales de la bomba de banda ancha. Por el contrario, con distancias focales más cortas, habrá más nm por mm en el plano del detector, reduciendo el espacio ocupado por la bomba dispersa.
La detección equilibrada mejora la calidad de imagen y la sensibilidad de las configuraciones ruidosas. Por ejemplo, de acuerdo con los espectros RIN que se muestran en la Figura 8 y considerando el SRS típico con una amplitud de 1 x 10-5, la relación señal-ruido (SNR) desequilibrada es de ≈60. Utilizando la detección equilibrada (es decir, cerca del ruido del disparo), es posible alcanzar un SNR de ≈145. La Figura 11 muestra espectros e imágenes compuestas en condiciones equilibradas y desequilibradas. Naturalmente, los efectos de la detección equilibrada afectan los resultados finales de los experimentos, es decir, los mapas químicos. Con el apoyo de estos resultados, enfatizamos que la detección equilibrada es una técnica poderosa para contrarrestar los efectos perjudiciales de las fluctuaciones del láser en la calidad de la imagen. Vale la pena mencionar que la detección equilibrada es más adecuada para láseres ruidosos, como los osciladores de fibra. Los microscopios SRS que funcionan con fuentes de luz óptica silenciosas (por ejemplo, láseres de estado sólido) pueden no requerir una detección equilibrada.
El protocolo también explica un enfoque basado en la óptica no lineal para encontrar la superposición espaciotemporal entre los pulsos de estos haces. Describimos las ventajas de usar el1º en lugar del0º orden de difracción de una OMA como el haz de Stokes modulado. Además, los efectos perjudiciales de la dispersión en la eficiencia de generación de SRS se describieron con sugerencias de formas de mitigarlos a través de un compresor prisma. Además, el protocolo explica cómo alinear los prismas y destaca tres aspectos críticos a considerar para un rendimiento óptimo. No solo discutimos la relevancia del RIN para la microscopía SRS, sino que también mostramos cómo medirlo con un amplificador de bloqueo y, con el espectro RIN, definir la mejor frecuencia de modulación. Con un ejemplo concreto, este documento explica cómo la ecuación de rejilla ayuda a diseñar la cadena de detección. Finalmente, el protocolo ilustra, con datos reales de SRS, la estructura del hipercubo SRS y cómo analizarlo con un lenguaje de programación científico utilizado convencionalmente.
Este protocolo tiene tres limitaciones menores. En primer lugar, el esquema de detección empleado en esta contribución consiste en un detector de bloqueo multicanal no convencional diseñado y construido internamente por Sciortino et al.26 Como se demostró anteriormente25, este detector puede ser reemplazado por un fotodiodo equilibrado listo para usar. Aunque esta modificación afecta solo al detector y deja el protocolo prácticamente sin cambios, con un solo fotodiodo, es necesario escanear cada componente espectral en el detector en lugar de medirlos todos a la vez. En segundo lugar, este protocolo emplea la detección equilibrada en línea, que requiere insertar varios elementos ópticos en la trayectoria del haz. Estos elementos ópticos aumentan la complejidad del sistema y conducen a pérdidas de potencia óptica y ampliación de pulsos.
La detección equilibrada en línea también exige que las dos réplicas de la bomba pasen a través de la muestra, una situación que puede no ser ideal para muestras sensibles a la luz, como las células vivas, o para las fuertemente birrefringentes en las que las dos réplicas de la bomba pueden experimentar diferentes propiedades ópticas, cancelando así la detección equilibrada. En tercer lugar, el protocolo se basa en un OPO construido en casa, un dispositivo que puede no estar fácilmente disponible. Sin embargo, las alternativas a los espectros de banda ancha entregados por el OPO son la supercontinua de fibras ópticas no lineales o cristales a granel. Este último podría emplearse solo con láseres de baja tasa de repetición (hasta 5 MHz). Por lo tanto, como con todo diseño experimental, el protocolo en cuestión tiene algunas limitaciones. Sin embargo, son mínimos y no comprometen el éxito de este enfoque.
Aunque aquí se describe una muestra de referencia, este protocolo puede desenredar con éxito especies químicas dentro de células y tejidos animales y vegetales, como celulosa, especies lipídicas o proteínas, encontrando aplicaciones prácticas en diferentes búsquedas bioquímicas o como herramienta de diagnóstico en histopatología. Del mismo modo, este protocolo puede ser una herramienta valiosa en las ciencias de los materiales. Por ejemplo, siguiendo este protocolo, se puede interrogar la composición molecular y la concentración de especies poliméricas42. Además, esta metodología es compatible con otras técnicas de microscopía no lineal, como la microscopía de banda ancha basada en bomba-sonda43 y la heterodina CARS44, procesos de mezcla de cuatro ondas que, al igual que con SRS, también requieren dos haces de luz de excitación y mediciones de modulación-transferencia. Finalmente, parte de la información contenida en este trabajo se puede aplicar a técnicas de imagen no lineal que no se basan en técnicas de transferencia de modulación, sino que requieren alinear dos o más rayos láser pulsados, como las microscopías convencionales CARS45 y SFG46.
En resumen, este protocolo describe una potente metodología basada en la microscopía SRS de banda ancha para extraer mapas químicos y sus espectros SRS característicos de mezclas químicamente heterogéneas, entregando conjuntos de datos que permiten un análisis de datos cuantitativo directo. La versatilidad y simplicidad del método también dan al lector interesado la posibilidad de adaptarlo a diferentes técnicas no lineales.
