The human body harbors an estimated 10-100 trillion live microbial cells (bacteria, archaea fungi), which are primarily found in the gut, skin, and mucosal environments1. In a healthy state, these provide benefits to their host, including vitamin production, maturation of the immune system, stimulation of innate and adaptive immune responses to pathogens, regulation of fat metabolism, modulation of stress responses, and more, with an impact on growth and development, disease onset, and ageing2,3,4,5. The gut microbiota also evolves considerably throughout life. The most drastic evolution occurs during infancy and early childhood6, but significant changes also occur with age, including a decrease in Bifidobacterium abundance and an increase in Clostridium, Lactobacillus, Enterobacteriaceae, and Enterococcus species7. Lifestyle can further alter gut microbial composition leading to dysbiosis (loss of beneficial bacteria, overgrowth of opportunistic bacteria), resulting in various pathologies such as inflammatory bowel disease, diabetes, and obesity5, but also contributing to Alzheimer's and Parkinson's diseases8,9,10,11.
This realization has critically contributed to refining the concept of the gut-brain axis (GBA), where interactions between gut physiology (now including the microbes within it) and the nervous system are considered the main regulator of animal metabolism and physiological functions12. However, the precise role of microbiota in gut-brain signaling and the associated mechanisms of action are far from being fully understood13. With gut microbiota being a key determinant of healthy aging, how bacteria modulate the aging process has become a subject of intense research and controversy6,14,15.
With the demonstration that the roundworm Caenorhabditis elegans hosts a bonafide gut microbiota dominated-as in other species-by Bacteroidetes, Firmicutes, and Actinobacteria16,17,18,19,20, its rapid rise as an experimental platform to study host-gut commensal interactions21,22,23,24,25,26 has significantly expanded our investigative arsenal26,27,28,29. In particular, high-throughput experimental approaches available for C. elegans to study gene-diet, gene-drug, gene-pathogen, etc. interactions, can be adapted to rapidly explore how bacterial isolates and cocktails impact C. elegans health and aging.
The present protocol describes an experimental pipeline to screen at once arrays of bacterial isolates or mixtures set in multiwell plates for effects on C. elegans stress resistance as a proxy for health, which can be used to identify probiotics. It details how to grow large worm populations and handle bacterial arrays in 96- and 384-well plate formats before processing worms for automated stress resistance analysis using a fluorescence plate reader (Figure 1). The approach is based on label-free automated survival assays (LFASS)30 that exploit the phenomenon of death fluorescence31, whereby dying worms produce a burst of blue fluorescence that can be used to pinpoint the time of death. Blue fluorescence is emitted by glucosyl esters of anthranilic acid stored in C. elegans gut granules (a type of lysosome-related organelle), which burst when a necrotic cascade is triggered in the worm gut upon death31.
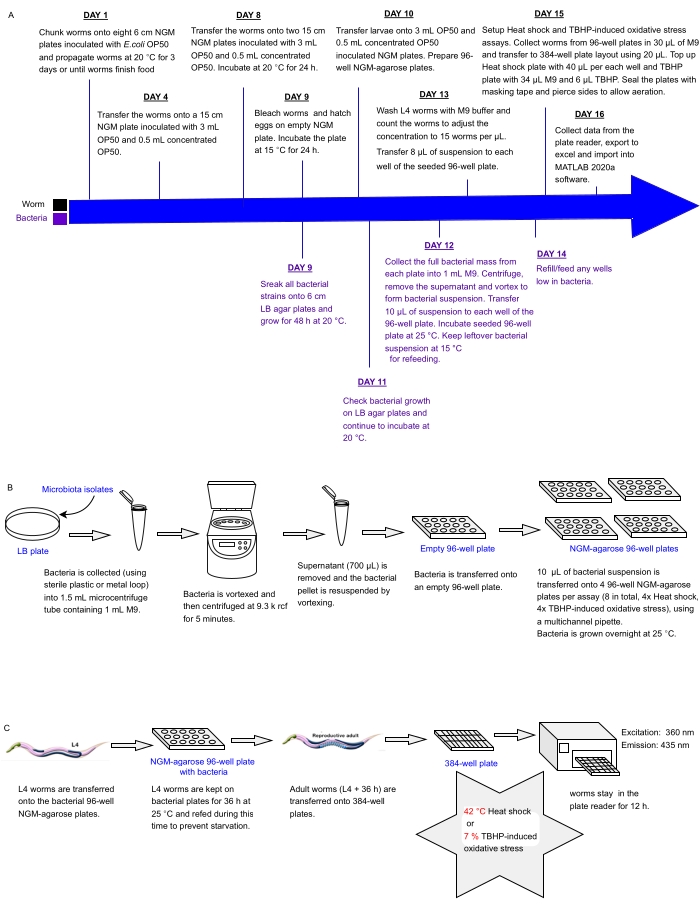
Figure 1: Experimental workflow for high-throughput screening of bacterial isolates with impact on C. elegans resistance to stress. (A) Timeline for worm and bacterial maintenance and assay setup. (B) 96-well bacterial plate array setup and handling. (C) 384-well worm plate setup. Please click here to view a larger version of this figure.
LFASS assays provide robust, high-throughput, and rapid screening of multiple test conditions at once, such as screening numerous genetic and microbiota parameters that contribute to stress resistance and aging. It only takes 2-3 weeks for the experiment to acquire an extensive dataset of multiple test conditions. L4 + 36 h adult wild-type worm populations were exposed to 42 °C thermal stress and 7% t-BHP-induced oxidative stress after a 36 h culture on 48 gut microbial isolates for 36 h. The assay was performed four times, with each condition replicated four times in each assay. Across all the conditions tested, the median time of death varied between 40-130 min for the thermal stress assay and between 90-240 min for the t-BHP induced oxidative stress assay. Early adulthood thermal stress assays usually display more consistent results and less inter-day and intra-day variability than oxidative stress assays and are better predictors of subsequent longevity30. The difference in the median time of death of worms fed on different microbial diets conclusively exemplifies how gut commensals impact host stress resistance. Figure 2 shows typical results from 16 biological replicates for Bristol N2 wild-type worms on two gut microbiota isolates of interest and the standard laboratory strain E. coli OP50.
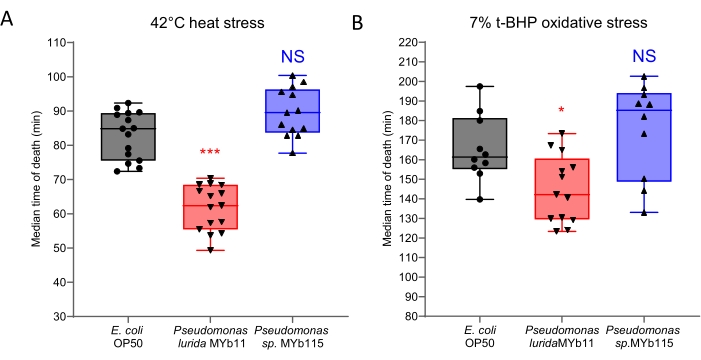
Figure 2: Representative results from paired heat/oxidative stress resistance assays from worms grown on two bacterial isolates and a control strain. (A) Heat stress assay. Wild-type worms were fed for 36 h on either MYb11 (Pseudomonas lurida) or MYb115 (Pseudomonassp.) bacterial isolates or OP50 control bacteria. MYb11 led to a significantly earlier median time of death (p < 0.001). (B) Oxidative stress assay. Wild-type worms were fed for 36 h on either MYb11 or MYb115 bacterial isolates or OP50 control bacteria. MYb11 led to a significantly earlier median time of death (p < 0.05). 120-150 worms were analyzed in four sets of samples, and each experiment was performed in quadruplicate. Wells with poor quality/poorly fitted data led to missing values. Box plot representations indicate single well values, minimum, median, and maximum values. *Indicates statistical significance at p < 0.05 and ***p < 0.001, with one-way ANOVA. Please click here to view a larger version of this figure.
| A | |||
| Parameter | Setting | Comment | |
| Temperature | 25 °C | ||
| Fluorescence excitation | 365 nm | 10 nm bandwidth | |
| Fluorescence emission | 430 nm | 20 nm bandwidth | |
| Number of flashes | 8 flashes, 1ms settle time | ||
| Gain | 100 to 130 | To be adjusted depending on sample. Aim for 2-5 k read value at start of assay | |
| Lag time | 1 μs | ||
| Integration time | 30 μs | ||
| Read time interval | Every 2 min | Depending on stress severity, duration and time interval may need adjusting (8 h is enough at 7% t-BHP) | |
| Duration | 8-12h | ||
| Read direction | From below | ||
| B | |||
| Parameter | Setting | Comment | |
| Temperature | 42 °C | ||
| Fluorescence excitation | 365 nm | 10 nm bandwidth | |
| Fluorescence emission | 430 nm | 20 nm bandwidth | |
| Number of flashes | 8 flashes, 1ms settle time | ||
| Gain | 100 to 130 | To be adjusted depending on sample. Aim for 2-5 k read value at start of assay | |
| Lag time | 1 μs | ||
| Integration time | 30 μs | ||
| Read time interval | Every 2 min | Depending on stress severity, duration and time interval may need adjusting (6 h is enough at 42 °C) | |
| Duration | 6-12h | ||
| Read direction | From below | ||
Table 1: Oxidative and heat stress assay settings. Examples of settings used for oxidative (A) and heat (B) stress assays on the fluorescence plate readers used in this study.
Supplementary File 1: Media, buffer, and culture recipes. Composition of different media, buffers, and cultures used in the present study. Please click here to download this File.
Supplementary File 2: Raw fluorescence data from the plate reader after being exported and converted to excel .xls format. (A) The excel file shows the raw fluorescence data for a heat shock assay. Raw fluorescence data is shown at 2 min time interval. (B) The column with "well position" on the 384-well plate is highlighted, which can be used to label the worm and bacterial strains. (C) The file is labeled with worm and bacterial strains. Please click here to download this File.
Supplementary File 3: LFASS analysis using Matlab. (A) After opening the LFASS folder in Matlab, a command window pops up. (B) "fitfolder" is typed in the command window to start the program, and the folder's name with the file to be analyzed is indicated. (C) Once the program finds the data excel file for analysis, the user must follow on-screen instructions, providing the various parameters requested. Please click here to download this File.
Supplemental File 4: Analyzed LFASS data using Matlab. (A) An example of a curve fitted through Matlab analysis, where the black line indicates the time point corresponding to the earlier time boundary and the blue line the later time boundary for the curve fitting. The fitted sigmoid curve is displayed in red. (B) The results file generated by Matlab is opened in .xls format. The values are highlighted and displayed in number format. The second column provides the time when the value on the raw data curve first exceeds 50% of the maximum value. The third column provides the time when the fitted curve's value equals 50% of the maximum value, as determined during the batch-fitting analysis. The fourth column provides the time at which the value on the fitted curve equals 50% of the maximum value after batch analysis and final refitting where necessary. The fourth column should, thus, provide the most reliable result. Please click here to download this File.
Supplementary Table 1: Example experimental plate layouts. (A) 96-well plate layout for testing 47 different worm gut microbiota bacterial isolates on two separate worm strains, with live OP50 being used as a control. Four copies of this plate are required for each assay being run. (B) 384-well layout for heat or oxidative stress assays, corresponding to the previous 96-well plate assay in (A). This setup allows tetraplicates to be performed for each condition, with each worm strain having its own OP50 control. Please click here to download this Table.
