Purificación y control de calidad de complejos de septina recombinante para la reconstitución libre de células
Summary
La reconstitución in vitro de proteínas citoesqueléticas es una herramienta vital para comprender las propiedades funcionales básicas de estas proteínas. El presente artículo describe un protocolo para purificar y evaluar la calidad de los complejos de septina recombinantes, que desempeñan un papel central en la división y migración celular.
Abstract
Las septinas son una familia de proteínas eucariotas conservadas de unión a GTP que pueden formar filamentos citoesqueléticos y estructuras de orden superior a partir de complejos heterooligoméricos. Interactúan con otros componentes citoesqueléticos y la membrana celular para participar en funciones celulares importantes como la migración y la división celular. Debido a la complejidad de las muchas interacciones de las septinas, el gran número de genes de septina (13 en humanos) y la capacidad de las septinas para formar complejos heterooligoméricos con diferentes composiciones de subunidades, la reconstitución libre de células es una estrategia vital para comprender los conceptos básicos de la biología de la septina. El presente artículo describe primero un método para purificar septinas recombinantes en su forma heterooligomérica utilizando un enfoque de cromatografía de afinidad de dos pasos. Luego, se detalla el proceso de control de calidad utilizado para verificar la pureza e integridad de los complejos de septina. Este proceso combina electroforesis en gel nativo y desnaturalizante, microscopía electrónica de tinción negativa y microscopía de dispersión interferométrica. Finalmente, se proporciona una descripción del proceso para verificar la capacidad de polimerización de los complejos de septina utilizando microscopía electrónica de tinción negativa y microscopía fluorescente. Esto demuestra que es posible producir hexámeros y octámeros de septina humana de alta calidad que contienen diferentes isoformas de septin_9, así como hexámeros de septina de Drosophila .
Introduction
El citoesqueleto se ha descrito clásicamente como un sistema de tres componentes que consiste en filamentos de actina, microtúbulos y filamentos intermedios 1, pero recientemente, las septinas han sido reconocidas como un cuarto componente del citoesqueleto1. Las septinas son una familia de proteínas de unión a GTP que se conservan en eucariotas2. Las septinas están involucradas en muchas funciones celulares como la división celular3, la adhesión célula-célula4, la motilidad celular5, la morfogénesis6, la infección celular7 y el establecimiento y mantenimiento de la polaridad celular8. A pesar de sus importantes funciones, la forma en que las septinas están involucradas en tales procesos es poco conocida.
La familia de proteínas de la septina se subdivide en varios subgrupos (cuatro o siete, dependiendo de la clasificación) basados en la similitud de la secuencia de proteínas2. Los miembros de diferentes subfamilias pueden formar complejos heterooligoméricos palindrómicos, que son los bloques de construcción de filamentos y que, a su vez, se ensamblan en estructuras de orden superior como haces, anillos y mallas 1,9,10,11,12. La complejidad molecular adicional surge de la presencia de diferentes variantes de empalme, un ejemplo es el SEPT9 humano, donde hay evidencia de funciones específicas de diferentes variantes de empalme13,14,15. Además, la longitud de los heterooligómeros depende de la especie y el tipo de célula. Por ejemplo, las septinas de Caenorhabditis elegans forman tetrámeros 16, las septinas de Drosophila melanogaster forman hexámeros 17 (Figura 1A), las septinas de Saccharomyces cerevisiae forman octameros 18 y las septinas humanas forman hexámeros y octámeros19 (Figura 1A). La capacidad de las isoformas de septina, las variantes de empalme y las septinas modificadas postraduccionalmente de la misma subfamilia para sustituirse entre sí en el complejo y la (co)existencia de heterooligómeros de diferentes tamaños han dificultado la delineación de las funciones celulares de diferentes complejos heterooligoméricos12.
Otra capacidad interesante de las septinas es su capacidad para interactuar con muchos socios de unión en la célula. Las septinas se unen a la membrana plasmática y a los orgánulos membranosos durante la interfase y la división celular20,21,22. En las células en división, las septinas cooperan con la anillina 23,24,25 y la actina y la miosina durante la citocinesis 26,27. En las últimas etapas de la citocinesis, las septinas parecen regular los complejos de clasificación endosomal requeridos para el sistema de transporte (ESCRT) para la abscisión del cuerpo medio28. Además, también hay evidencia de septina localizada en la corteza de actina y fibras de estrés de actina de las células en las células de interfase 29,30,31. En tipos celulares específicos, las septinas también se unen y regulan el citoesqueleto de microtúbulos32,33.
Todas estas características hacen que las septinas sean un sistema proteico muy interesante para estudiar, pero también desafiante. La combinación del gran número de subunidades de septina (13 genes en humanos sin contar las variantes de empalme2) con el potencial de las subunidades de septina de la misma subfamilia para sustituirse entre sí y formar heterooligómeros de diferentes tamaños hace que sea difícil sacar una conclusión sobre la función celular de una septina específica por manipulación genética. Además, las múltiples interacciones de las septinas hacen que interpretar los efectos de herramientas de investigación comunes, como los medicamentos34 dirigidos a los componentes citoesqueléticos o de membrana, sea una tarea difícil.
Una forma de superar esta situación es complementar la investigación en células con la reconstitución in vitro (libre de células) de septinas. La reconstitución in vitro permite el aislamiento de un solo tipo de heterooligómeros de septina con una composición de subunidad específica y longitud 18,35,36,37. Este complejo puede ser estudiado en un ambiente controlado, ya sea solo para descubrir las propiedades estructurales y fisicoquímicas básicas de las septinas 38,39,40, o en combinación con socios deseados como biomembranas modelo 11,41,42, filamentos de actina 10,27 o microtúbulos 32,36 para descifrar la naturaleza de sus Interacciones.
Por lo tanto, un método confiable para purificar diferentes complejos de septina de manera eficiente es vital para la investigación de septinas. Sin embargo, incluso usando el mismo protocolo, diferentes purificaciones pueden dar proteínas con diferente actividad / funcionalidad o incluso integridad. Para las proteínas disponibles comercialmente, como las enzimas, la funcionalidad y la actividad enzimática se validan cuidadosamente43. Implementar un control de calidad cuidadoso para las proteínas citoesqueléticas o estructurales como las septinas puede ser un desafío, pero es esencial hacer que los experimentos entre laboratorios sean comparables.
Este artículo describe un método robusto para purificar septinas recombinantes de alta calidad en su forma heterooligomérica basado en la expresión simultánea de dos vectores que contienen construcciones mono o bicistrónicas (Tabla 1) en células de Escherichia coli. El método consiste en un enfoque de cromatografía de afinidad de dos pasos para capturar heterooligómeros de septina que contienen tanto una septina marcada con his6 como una septina marcada con Strep-II (Figura 1B, C). Este protocolo, descrito por primera vez en Iv et al.10, se ha utilizado para purificar hexámeros de septina 11,27,35 de Drosophila, hexámeros de septina humana 10 y varios octámeros de septina humanos que contienen diferentes isoformas nativas (isoformas 1, 3 y 5)10,32 o SEPT9 mutadas 32 . Además, se describe un conjunto de técnicas para evaluar la calidad de las septinas purificadas. En primer lugar, se comprueba la integridad y la estequiometría correcta de las subunidades de septina mediante electroforesis desnaturalizante y microscopía electrónica de transmisión (TEM). Luego, la presencia de heterooligómeros de la masa molecular correcta y la presencia de monómeros u oligómeros más pequeños indicativos de inestabilidad compleja se examinan mediante electroforesis nativa y fotometría de masas mediante microscopía de dispersión interferométrica (iSCAT). Finalmente, el último paso consiste en la evaluación de la actividad polimerizante de las septinas mediante microscopía de fluorescencia y TEM.
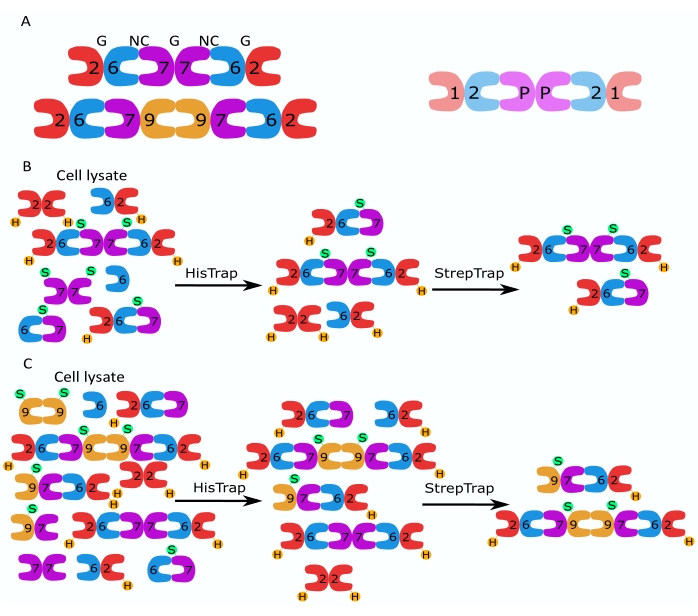
Figura 1: Estrategia de purificación. (A) Esquemas de los heterooligómeros de septina que existen en células humanas (izquierda) y Drosophila (derecha). Los números denotan subunidades de septina de los grupos indicados, y P denota maní. El SEPT9 humano puede ser cualquiera de sus isoformas. Las subunidades de septina tienen una forma asimétrica y están asociadas longitudinalmente con dos interfaces distintas, la interfaz NC: NC y la interfaz G: G, como se indica por NC y G, respectivamente, en la parte superior del hexámero humano. (B,C) Ilustración esquemática de la estrategia de cromatografía en dos pasos, mostrada para (B) hexámeros de septina humanos y (C) octamers. H indica las etiquetas his, mientras que S indica las etiquetas Strep-II. Haga clic aquí para ver una versión más grande de esta figura.
Protocol
Representative Results
Discussion
El método descrito aquí permite la purificación robusta y el control de calidad de heterooligómeros de septina preformados. Algunas de las cuestiones clave a considerar para la correcta aplicación del método son las siguientes. Durante los pasos de elución en las separaciones cromatográficas, es importante utilizar el caudal recomendado (o inferior) para minimizar la dilución de los complejos de septina. Además, para maximizar la recuperación de proteínas durante el paso final de concentración, la columna del concentrador está orientada de tal manera que la solución no se empuja contra el filtro (cuando solo hay un filtro en un lado). Si la solución va directamente al filtro, la proteína se adhiere mucho más a él, disminuyendo sustancialmente el rendimiento final. También es importante considerar que el paso de concentración no siempre es necesario. La selección de fracciones solo de un rango estrecho alrededor del pico en el cromatograma generalmente da una concentración de stock suficientemente alta (>3,000 nM) para muchas aplicaciones de reconstitución (que generalmente operan entre 10-300 nM). Finalmente, para el control de calidad de la funcionalidad de los complejos de septina por microscopía de fluorescencia, es importante pasivar correctamente la superficie de los portaobjetos de microscopía, ya que los complejos de septina se adhieren ávidamente al vidrio. La pasivación de los portaobjetos de vidrio se puede realizar mediante funcionalización PLL-PEG o mediante la formación de bicapas lipídicas neutras (100% DOPC) soportadas11,32.
En comparación con el protocolo de purificación original descrito por primera vez en Iv et al.10, hay un cambio en las composiciones de los tampones (Tabla 2). La concentración de MgCl 2 se ha reducido de 5 mM a2 mM, y la concentración y el pH de Tris-HCl se han reducido de 50 mM a 20 mM y de 8,0 a 7,4, respectivamente. Estos cambios se realizaron para hacer compatibles las condiciones de amortiguación con los estudios de las interacciones de las septinas humanas con bicapas lipídicas, filamentos de actina y microtúbulos10,11,32. Esto se debe a que los autores formaron bicapas lipídicas soportadas y actina polimerizada en el F-buffer, cuya composición es idéntica a la de darkSPB, aparte de la presencia de ATP en el F-buffer. El cambio de tampón no produjo ningún cambio en la calidad o vida útil de las septinas purificadas en comparación con los tampones originales.
Este método de purificación todavía tiene varias limitaciones. En primer lugar, los diferentes intentos de purificación pueden variar en rendimiento (0,5-1 ml de complejos de septina de 2-5 μM) y calidad funcional, como lo comprueba la capacidad de formación del haz de los complejos de septina purificados. Es por eso que es muy importante realizar consistentemente los controles de calidad descritos en este documento. Controlar muy bien los tiempos de expresión y la densidad óptica del cultivo bacteriano puede ayudar a mitigar la diferencia en el rendimiento. En segundo lugar, esta tubería de purificación no puede distinguir entre trímeros y hexámeros o entre tetrámeros y octámeros (Figura 1B). Sin embargo, los experimentos de control de calidad se pueden utilizar para demostrar que la mayoría de los complejos de septina están en su forma de oligómero largo. En caso de que se requiera una distribución de tamaño de oligómero aún más estrecha, se puede insertar cromatografía de exclusión de tamaño entre el paso 1.6. y paso 1.7. del protocolo de purificación. Este paso opcional, sin embargo, disminuye drásticamente el rendimiento, y no se recomienda a menos que sea estrictamente necesario. Una última limitación, más fundamental, proviene del uso de E. coli como sistema de expresión para complejos de septina recombinantes. Naturalmente, este sistema no permite modificaciones postraduccionales (PTM), que han sido reportadas en células animales, como fosforilación, acetilación y sumoilación 6,51,52,53. Estas modificaciones postraduccionales podrían agregarse implementando una estrategia de purificación similar en células de insectos o humanas. Además, este documento solo ha discutido la reconstitución de septinas por sí mismas, pero los estudios en células indican que las proteínas reguladoras como las proteínas de la familia Borg 54,55 y la anillina 24,25,56 pueden tener efectos sustanciales pero poco conocidos sobre el ensamblaje y las funciones de las septinas y, por lo tanto, son importantes para incorporarlas eventualmente in vitro. Estudios. Los protocolos para la purificación de las proteínas Borg y la anillina han sido reportados54,57.
El protocolo de purificación de septina reportado aquí ofrece una forma estandarizada de purificar septinas en su forma de oligómero con la estequiometría de subunidades correcta, ofreciendo un avance importante sobre muchos estudios in vitro anteriores basados en subunidades de septina individuales. A pesar de que algunas septinas en contextos específicos pueden actuar como una sola subunidad2, el cuerpo actual de literatura sugiere fuertemente que, en las células animales, las septinas funcionan principalmente en complejos 9,58. Por lo tanto, el uso de heterooligómeros preformados, como los descritos en este trabajo y otros 10,11,18,32,35,36,37, es de gran importancia para estudiar las propiedades estructurales y biofísicas de las septinas vía in vitro. reconstitución para diseccionar sus funciones en la célula. Además, las septinas son proteínas autoensamblables con muchos socios de interacción, incluyendo la membrana y el citoesqueleto, lo que las hace de gran interés para la biología sintética ascendente 59,60,61 y estudios de cambios inducidos por proteínas en las propiedades biofísicas de la membrana como la curvatura42,62,63.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Agradecemos a Cecilia de Agrela Pinto, Tomás de Garay y Katharina Häußermann por su ayuda con los experimentos de fotometría de masas (iSCAT); Arjen Jakobi y Wiel Evers por su ayuda con TEM; Lucia Baldauf por su asistencia con TIRF; Pascal Verdier-Pinard por sus consejos sobre la electroforesis nativa; Agata Szuba y Marjolein Vinkenoog por su ayuda en la creación de los esfuerzos de purificación de septina de Drosophila, y el Cell and Tissue Imaging (PICT-IBiSA), Institut Curie, miembro de la Infraestructura Nacional de Investigación de Francia-BioImaging (ANR10-INBS-04). Esta investigación recibió financiación de la Organización Holandesa para la Investigación Científica (NWO/OCW) a través de la subvención de gravitación ‘BaSyC-Building a Synthetic Cell’ (024.003.019) y de la Agence Nationale pour la Recherche (ANR grants ANR-17-CE13-0014: “SEPTIMORF”; ANR-13-JSV8-0002-01: “SEPTIME”; y ANR-20-CE11-0014-01: “SEPTSCORT”).
Materials
| 488nm laser combiner iLAS2 | Gataca | TIRF microscope | |
| 488nm Sapphire laser lines | Coherent | Confocal microscope | |
| 4k X 4k F416 CMOS camera | TVIPS | For JEM-1400plus | |
| 4x sample buffer nativePAGE | Thermo Fisher scientific | BN2003 | |
| 6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (TROLOX) | Sigma-Aldrich | 238813 | To prevent blinking |
| AKTA pure 25 M1 | GE healthcare | 1680311 | |
| Ampicillin | Sigma-Aldrich | A9518-25G | |
| Carbon Type-B, 300 mesh EM grid | Ted pella | 01813-F | |
| Carbon Type-B, 300 mesh EM grid | Electron micoscopy sciences | CF300-Cu | |
| Cover glass #1.5H | Thorslabs | CG15KH | |
| CSU-X1-M1 confocal unit | Yokogawa | Confocal microscope | |
| Desthiobiotin | Sigma-Aldrich | D1411-1G | |
| Dithiothreitol (DTT) | Sigma-Aldrich | D9779 | |
| DNAse | Sigma-Aldrich | 10104159001 | |
| DOPC | Avanti Polar Lipids | 850375C | |
| Eclipse Ti2-E | Nikon instruments | Confocal microscope | |
| EDTA-free protease inhibtor cocktail | Roche | 481761 | |
| HisTrap HP, 5 mL | GE healthcare | 29-0588-3 | |
| iLAS2 azimuthal TIRF illumination system | Gataca | TIRF microscope | |
| Imidazole | Sigma-Aldrich | 1202-1KG | |
| InstantBlue Protein Gel Stain | Westburg Life Sciences | EP ab119211 | |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Thermo Fisher scientific | 10849040 | |
| iXon Ultra 888 EMCCD camera | Andor | Confocal microscope | |
| iXon Ultra 897 EM-CCD | Andor | TIRF microscope | |
| JEM-1400plus | JOEL | TEM microscope TUDelft | |
| kappa-cassein | Sigma-Aldrich | C0406 | |
| LB broth | Sigma-Aldrich | L3022-6X1KG | |
| Lyzozyme | Sigma-Aldrich | 62971-10G-F | |
| Magnesium Chloride | Sigma-Aldrich | M8266-100G | |
| Magnesium sulfate | Sigma-Aldrich | 746452-1KG | |
| Methylecllulose | Sigma-Aldrich | 8074844 | |
| MilliQ system (Integral 10) | Merck-Millipore | I-water dispenser | |
| Mini protean TGX gels | BIORAD | 4561086 | |
| NativeMark unstained protein standard | Invitrogen | LC0725 | For iSCAT and Native gels |
| NativePAGE 4-16% GELS | Thermo Fisher scientific | BN1002BOX | |
| NativePAGE Running Buffer kit | Thermo Fisher scientific | BN2007 | |
| Nikon Ti2-E | Nikon instruments | TIRF microscope | |
| Nr. 1 Menzel coverslips | Thermo Fisher scientific | 11961988 | |
| parafilm | Sigma-Aldrich | P7668 | |
| Plan Apo ×100/1.45 NA oil immersion objective | Nikon instruments | Confocal microscope | |
| PMSF | Sigma-Aldrich | 10837091001 | |
| Poly(L-lysine)-graft-biotinylated PEG (PLL-PEG) | SuSoS | CHF560.00 | |
| Poly-L-lysine solution 0.01% | Sigma-Aldrich | P4832 | For iSCAT glass slides |
| Pottassium Chloride | Sigma-Aldrich | P9541-1KG | |
| Power supply for native gels | CONSORT | S/N 71638 | |
| POWERPAC UNIVERSAL | BIORAD | 042BR31206 | |
| Protocatechuate 3,4-Dioxygenase (PCD) | Sigma-Aldrich | P8279-25UN | oxygen scavenger – enzyme |
| Protocatechuic acid (PCA) | Sigma-Aldrich | 03930590-50MG | oxygen scavenger – reagent |
| Q500 Sonicator | Qsonica | Q500-110 | |
| Quemesa camera | Olympus | For Tecnai Spirit | |
| Refeyn OneMP | Refeyn | ||
| Sample buffer, laemmli 2x concentrate | Sigma-Aldrich | S3401-10vl | |
| Silicon gaskets | Sigma-Aldrich | GBL103250-10EA | |
| Slide-A-Lyzer Dialysis cassettes 30k MWCO 3mL | Thermo Fisher scientific | 66381 | |
| Spectinomycin | Sigma-Aldrich | PHR1441-1G | |
| StrepTrap HP, 1 mL | GE healthcare | 28-9075-46 | |
| Tecnai Spirit microscope | Thermo Scientific, FEI | TEM microscope Institute Curie | |
| Terrific broth | Sigma-Aldrich | T0918-1KG | |
| Tris/Glyine/SDS buffer | BIORAD | 1610772 | |
| Tris-HCl | Sigma-Aldrich | T5941-1KG | |
| Ultrasonic cleaner | Branson | CPX2800H-E | |
| Vivaspin 6, 30,000 MWCO PES | Sartorius | VS0622 |
References
- Mostowy, S., Cossart, P. Septins: The fourth component of the cytoskeleton. Nature Reviews Molecular Cell Biology. 13 (3), 183-194 (2012).
- Shuman, B., Momany, M. Septins from protists to people. Frontiers in Cell and Developmental Biology. 9, 3802 (2022).
- Bridges, A. A., Gladfelter, A. S. Septin form and function at the cell cortex. Journal of Biological Chemistry. 290 (28), 17173-17180 (2015).
- Smith, C., et al. Septin 9 exhibits polymorphic binding to F-actin and inhibits myosin and cofilin activity. Journal of Molecular Biology. 427 (20), 3273-3284 (2015).
- Gilden, J. K., Peck, S., Chen, Y. C. M., Krummel, M. F. The septin cytoskeleton facilitates membrane retraction during motility and blebbing. Journal of Cell Biology. 196 (1), 103-114 (2012).
- Marquardt, J., Chen, X., Bi, E. Architecture, remodeling, and functions of the septin cytoskeleton. Cytoskeleton. 76 (1), 7-14 (2018).
- Van Ngo, H., Mostowy, S. Role of septins in microbial infection. Journal of Cell Science. 132 (9), (2019).
- Fung, K. Y. Y., Dai, L., Trimble, W. S. Cell and molecular biology of septins. International Review of Cell and Molecular Biology. 310, 289-339 (2014).
- Kinoshita, M., Field, C. M., Coughlin, M. L., Straight, A. F., Mitchison, T. J. Self- and actin-templated assembly of mammalian septins. Developmental Cell. 3 (6), 791-802 (2002).
- Iv, F., et al. Insights into animal septins using recombinant human septin octamers 2 with distinct SEPT9 isoforms. Journal of Cell Science. 134 (15), (2021).
- Szuba, A., et al. Membrane binding controls ordered self-assembly of animal septins. eLife. 10, 63349 (2021).
- Kinoshita, M. Assembly of mammalian septins. Journal of Biochemistry. 134 (4), 491-496 (2003).
- Connolly, D., et al. Septin 9 isoform expression, localization and epigenetic changes during human and mouse breast cancer progression. Breast Cancer Research. 13 (4), 76 (2011).
- Connolly, D., et al. Septin 9 amplification and isoform-specific expression in peritumoral and tumor breast tissue. Biological Chemistry. 395 (2), 157-167 (2014).
- Estey, M. P., Di Ciano-Oliveira, C., Froese, C. D., Bejide, M. T., Trimble, W. S. Distinct roles of septins in cytokinesis: SEPT9 mediates midbody abscission. Journal of Cell Biology. 191 (4), 741-749 (2010).
- John, C. M., et al. The Caenorhabditis elegans septin complex is nonpolar. EMBO Journal. 26 (14), 3296-3307 (2007).
- Field, C. M., et al. A purified Drosophila septin complex forms filaments and exhibits GTPase activity. Journal of Cell Biology. 133 (3), 605-616 (1996).
- Bertin, A., et al. Saccharomyces cerevisiae septins: Supramolecular organization of heterooligomers and the mechanism of filament assembly. Proceedings of the National Academy of Sciences of the United States of America. 105 (24), 8274-8279 (2008).
- Sellin, M. E., Sandblad, L., Stenmark, S., Gullberg, M. Deciphering the rules governing assembly order of mammalian septin complexes. Molecular Biology of the Cell. 22 (17), 3152-3164 (2011).
- Akil, A., et al. Septin 9 induces lipid droplets growth by a phosphatidylinositol-5-phosphate and microtubule-dependent mechanism hijacked by HCV. Nature Communications. 7, 12203 (2016).
- Tanaka-Takiguchi, Y., Kinoshita, M., Takiguchi, K. Septin-mediated uniform bracing of phospholipid membranes. Current Biology. 19 (2), 140-145 (2009).
- Omrane, M., et al. Septin 9 has two polybasic domains critical to septin filament assembly and Golgi integrity. iScience. 13, 138-153 (2019).
- Carim, S. C., Kechad, A., Hickson, G. R. X. Animal cell cytokinesis: The rho-dependent actomyosin-anilloseptin contractile ring as a membrane microdomain gathering, compressing, and sorting machine. Frontiers in Cell and Developmental Biology. 8, 575226 (2020).
- El Amine, N., Kechad, A., Jananji, S., Hickson, G. R. X. Opposing actions of septins and Sticky on Anillin promote the transition from contractile to midbody ring. Journal of Cell Biology. 203 (3), 487-504 (2013).
- Renshaw, M. J., Liu, J., Lavoie, B. D., Wilde, A. Anillin-dependent organization of septin filaments promotes intercellular bridge elongation and Chmp4B targeting to the abscission site. Open Biology. 4 (1), 130190 (2014).
- Vogt, E. T., et al. The ultrastructural organization of actin and myosin II filaments in the contractile ring: new support for an old model of cytokinesis. Molecular Biology of the Cell. 28 (5), 613-623 (2017).
- Mavrakis, M., et al. Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nature Cell Biology. 16 (4), 322-334 (2014).
- Karasmanis, E. P., et al. A septin double ring controls the spatiotemporal organization of the ESCRT machinery in cytokinetic abscission. Current Biology. 29 (13), 2174-2182 (2019).
- Hagiwara, A., et al. Submembranous septins as relatively stable components of actin-based membrane skeleton. Cytoskeleton. 68 (9), 512-525 (2011).
- Calvo, F., et al. Cdc42EP3/BORG2 and septin network enables mechano-transduction and the emergence of cancer-associated fibroblasts. Cell Reports. 13 (12), 2699-2714 (2015).
- Salameh, J., Cantaloube, I., Benoit, B., Poüs, C., Baillet, A. Cdc42 and its BORG2 and BORG3 effectors control the subcellular localization of septins between actin stress fibers and microtubules. Current Biology. 31 (18), 4088-4103 (2021).
- Kuzmić, M., et al. Septin-microtubule association via a motif unique to isoform 1 of septin 9 tunes stress fibers. Journal of Cell Science. 135 (1), (2022).
- Shindo, A., et al. Septin-dependent remodeling of cortical microtubule drives cell reshaping during epithelial wound healing. Journal of Cell Science. 131 (12), (2018).
- Hu, Q., Nelson, W. J., Spiliotis, E. T. Forchlorfenuron alters mammalian septin assembly, organization, and dynamics. Journal of Biological Chemistry. 283 (43), 29563-29571 (2008).
- Mavrakis, M., Tsai, F. C., Koenderink, G. H. Purification of recombinant human and Drosophila septin hexamers for TIRF assays of actin-septin filament assembly. Methods in Cell Biology. 136, 199-220 (2016).
- Nakos, K., Radler, M. R., Spiliotis, E. T. Septin 2/6/7 complexes tune microtubule plus-end growth and EB1 binding in a concentration- and filament-dependent manner. Molecular Biology of the Cell. 30 (23), 2913-2928 (2019).
- Kaplan, C., et al. Absolute arrangement of subunits in cytoskeletal septin filaments in cells measured by fluorescence microscopy. Nano Letters. 15 (6), 3859-3864 (2015).
- Castro, D. K. S. V., et al. A complete compendium of crystal structures for the human SEPT3 subgroup reveals functional plasticity at a specific septin interface. IUCrJ. 7, 462-479 (2020).
- Jiao, F., Cannon, K. S., Lin, Y. -. C., Gladfelter, A. S., Scheuring, S. The hierarchical assembly of septins revealed by high-speed AFM. Nature Communications. 11 (1), 1-13 (2020).
- Bertin, A., et al. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. Journal of Molecular Biology. 404 (4), 711-731 (2010).
- Bridges, A. A., Jentzsch, M. S., Oakes, P. W., Occhipinti, P., Gladfelter, A. S. Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. Journal of Cell Biology. 213 (1), 23-32 (2016).
- Beber, A., et al. Membrane reshaping by micrometric curvature sensitive septin filaments. Nature Communications. 10, 420 (2019).
- Zhou, R., Shi, Y., Yang, G. Expression, purification, and enzymatic characterization of intramembrane proteases. Methods in Enzymology. 584, 127-155 (2017).
- Diebold, M. L., Fribourg, S., Koch, M., Metzger, T., Romier, C. Deciphering correct strategies for multiprotein complex assembly by co-expression: Application to complexes as large as the histone octamer. Journal of Structural Biology. 175 (2), 178-188 (2011).
- Lebedeva, M. A., Palmieri, E., Kukura, P., Fletcher, S. P. Emergence and rearrangement of dynamic supramolecular aggregates visualized by interferometric scattering microscopy. ACS Nano. 14 (9), 11160-11168 (2020).
- Ludtke, S. J., Baldwin, P. R., Chiu, W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. Journal of Structural Biology. 128 (1), 82-97 (1999).
- Zivanov, J., et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 7, 42166 (2018).
- Frank, J., et al. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. Journal of Structural Biology. 116 (1), 190-199 (1996).
- Young, G., Kukura, P. Interferometric scattering microscopy. Annual Review of Physical Chemistry. 70, 301-322 (2019).
- Young, G., et al. Quantitative mass imaging of single biological macromolecules. Science. 360 (6387), 423-427 (2018).
- Hernández-Rodríguez, Y., Momany, M. Posttranslational modifications and assembly of septin heteropolymers and higher-order structures. Current Opinion in Microbiology. 15 (6), 660-668 (2012).
- Ribet, D., et al. SUMOylation of human septins is critical for septin filament bundling and cytokinesis. Journal of Cell Biology. 216 (12), 4041-4052 (2017).
- Sinha, I., et al. Cyclin-dependent kinases control septin phosphorylation in Candida albicans hyphal development. Developmental Cell. 13 (3), 421-432 (2007).
- Sheffield, P. J., et al. Borg/Septin interactions and the assembly of mammalian septin heterodimers, trimers, and filaments. Journal of Biological Chemistry. 278 (5), 3483-3488 (2003).
- Joberty, G., et al. Borg proteins control septin organization and are negatively regulated by Cdc42. Nature Cell Biology. 3 (10), 861-866 (2001).
- Chen, X., Wang, K., Svitkina, T., Bi, E. Critical roles of a RhoGEF-anillin module in septin architectural remodeling during cytokinesis. Current Biology. 30 (8), 1477-1490 (2020).
- Kučera, O., et al. Anillin propels myosin-independent constriction of actin rings. Nature Communications. 12 (1), 1-12 (2021).
- Hsu, S. C., et al. Subunit composition, protein interactions, and structures of the mammalian brain sec6/8 complex and septin filaments. Neuron. 20 (6), 1111-1122 (1998).
- Olivi, L., et al. Towards a synthetic cell cycle. Nature Communications. 12 (1), 1-11 (2021).
- Hürtgen, D., Härtel, T., Murray, S. M., Sourjik, V., Schwille, P. Functional modules of minimal cell division for synthetic biology. Advanced Biosystems. 3 (6), 1800315 (2019).
- Jia, H., Schwille, P. Bottom-up synthetic biology: Reconstitution in space and time. Current Opinion in Biotechnology. 60, 179-187 (2019).
- Cannon, K. S., Woods, B. L., Crutchley, J. M., Gladfelter, A. S. An amphipathic helix enables septins to sense micrometer-scale membrane curvature. The Journal of Cell Biology. 218 (4), 1128-1137 (2019).
- Lobato-Márquez, D., Mostowy, S. Septins recognize micron-scale membrane curvature. Journal of Cell Biology. 213 (1), 5-6 (2016).
