Over the last few years, the increased prevalence of obesity and T2D has prompted an intense search for the mechanisms mediating insulin resistance in adipose tissue. With the protocol described here, APCs can be differentiated into mature adipocytes to evaluate insulin resistance and sensitivity. Once the APCs reach confluence, it takes 10 days to complete their differentiation into mature adipocytes and their TNF-α-mediated induction of insulin resistance (Figure 1).
APCs show a fibroblast-like morphology, characterized by their flat and elongated shape and their adhesion to the plate completed 48 h after seeding (Figure 2A). APCs take 2-5 days to reach 80% confluence (depending on the number of seeded cells), a time at which the differentiation process is started by exposure to the differentiating cocktail (Figure 2A). Mature adipocytes begin to appear in the following 6-8 days. Differentiated adipocytes are characterized by a round morphology and the intracellular accumulation of lipid droplets. Up to 80% of cells are differentiated at the end of the differentiation process (Figure 2B).
Insulin resistance is induced in primary adipocytes by TNF-α treatment (4 ng/mL) for 48 h; this concentration of TNF-α does not alter cell viability (Supplementary Figure S3). Subsequently, the insulin signaling pathway is activated by adding 100 nM insulin for 15 min, and the phosphorylation of main signaling molecules (IR, IRS-1, and AKT) is measured by western blot. Insulin stimulates the phosphorylation of these three molecules (Figure 3) compared to control, non-treated differentiated adipocytes. TNF-α reduces the insulin-induced phosphorylation of IR (30%), IRS-1 (20%), and AKT (45%) (Figure 3). Insulin-resistant cells show less activation of the insulin signaling pathway compared to insulin-sensitive cells. Moreover, TNF-α treatment decreases the mRNA expression of known insulin-sensitivity markers: Insr, Irs2, Glut4, and Adipoq (Supplementary Figure S4). These findings confirm that TNF-α reduces the action of insulin in primary adipocytes, and thereby induces insulin resistance.
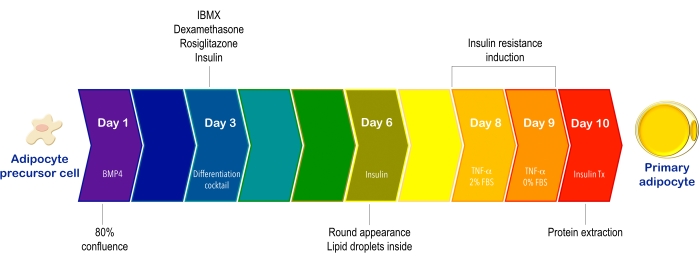
Figure 1: Schematic timeline representing the process of adipocyte precursor cell differentiation and the induction of insulin resistance. APCs are isolated from subcutaneous adipose tissue using collagenase treatment and magnetic cell separation technology. Then, they undergo a 7 day differentiation process to obtain primary adipocytes. Subsequently, insulin resistance is induced with TNF-α for 48 h, the first 24 h in medium containing 2% FBS and the next 24 h with serum-free medium to prevent the activation of the insulin signaling pathway. The signaling cascade is activated with insulin and protein is extracted for western blot analysis 10 days after starting the differentiation process to evaluate the phosphorylation/activation of IR, IRS-1, and AKT. Abbreviations: APCs = adipocyte precursor cells; BMP4 = Bone morphogenetic protein; IBMX = 3-isobutyl-1-methylxanthine; TNF-α = tumor necrosis factor-α; FBS = fetal bovine serum; IR = insulin receptor; IRS = insulin receptor substrate; AKT = protein kinase B; Tx = treatment. Please click here to view a larger version of this figure.
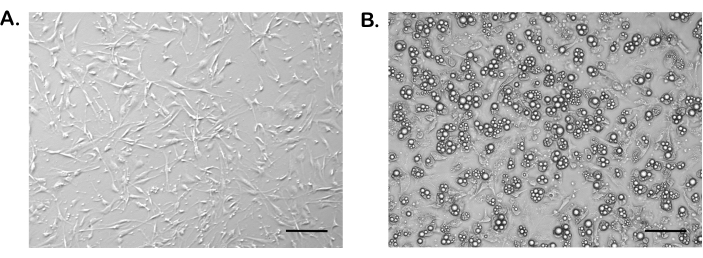
Figure 2: Morphology of adipocyte precursor cells and mature adipocytes. (A) Subcutaneous APCs at 80% confluence prior to inducing differentiation in 12-well culture plates. (B) Subcutaneous primary adipocytes after 7 days of inducing differentiation in 12-well culture plates. Images were taken at 10x magnification. Scale bars = 100 µm. Abbreviation: APCs = adipocyte precursor cells. Please click here to view a larger version of this figure.
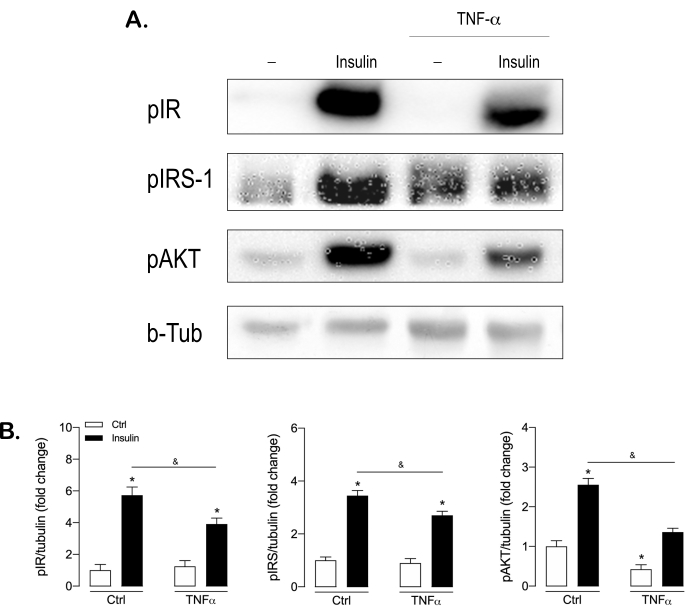
Figure 3: Insulin resistance induced by TNF-α in subcutaneous primary adipocytes demonstrated by decreased insulin-induced phosphorylation of IR, IRS-1, and AKT. Subcutaneous adipocytes were treated with TNF-α (4 ng/mL) for 48 h and serum-starved for the last 24 h. Following the stimulation with insulin (100 nM) for 15 min, 40 µg of total protein was loaded on 7.5% gels and subjected to SDS-PAGE, transferred to nitrocellulose membranes, and blocked in 4% non-fat milk in TBS-T 0.1%. The membrane was probed with anti-phosphorylated IR (pIR), anti-phosphorylated IRS-1 (pIRS-1), and anti-phosphorylated AKT (pAKT), as well as with an anti-rabbit HRP secondary antibody. The signal was visualized with chemiluminescence detection and anti-β tubulin was used as a loading control. (A) Representative blots and (B) quantification from three independent experiments. Data are mean ± SEM; *, p < .05 versus control. Abbreviations: TNF-α = tumor necrosis factor-α; IR = insulin receptor; IRS = insulin receptor substrate; pX = phosphorylated form of X; b-Tub = beta-tubulin; HRP = horseradish peroxidase; Ctrl = control. Please click here to view a larger version of this figure.
| Anti-Fc solution | purified rat anti-mouse CD16 / CD32 diluted in PBS–2% FBS [1:150] | |||
| TBS-T 0.1% | 0.01 M Tris-HCl (pH 8), 0.15 M NaCl, 0.1% Tween 20 | |||
| Blocking solution | 4% nonfat dry milk diluted in TBS-T 0.1% | |||
| Growth medium | 60% DMEM low glucose, 40% MCDB 201 medium, 1x penicillin-streptomycin, 1 nM dexamethasone, 0.1 mM L-ascorbic acid 2-phosphate, 1x insulin, transferrin, sodium, selenite (ITS) liquid media supplement, 1x linoleic acid-albumin from BSA, 10% FBS, 10 ng/mL epidermal growth factor (EGF), 10 ng/mL leukemia inhibitory factor (LIF), 10 ng/mL platelet-derived growth factor BB (PDGF-BB), 5 ng/mL fibroblast growth factor-basic (bFGF), and 50 µg/mL normocin | |||
| Differentiation medium | 60% DMEM low glucose, 40% MCDB 201 medium, 1x penicillin-streptomycin, 1 nM dexamethasone, 0.1 mM L-ascorbic acid 2-phosphate, 1x ITS liquid media supplement, 1x linoleic acid-albumin from BSA, and 2% FBS | |||
| Differentiation cocktail | 0.5 µM 3-isobutyl-1-methylxanthine [IBMX], 1 µM dexamethasone, 10 µM rosiglitazone, and 100 nM insulin | |||
| Simple medium-2% FBS | 60% DMEM low glucose, 40% MCDB 201 medium, 1x penicillin-streptomycin, and 2% FBS | |||
| Simple medium–0% FBS | 60% DMEM low glucose, 40% MCDB 201 medium, 1x penicillin-streptomycin | |||
| RIPA buffer | 50 mM Tris-HCl, 1 mM EGTA, 1 mM EDTA, 1% octylphenoxy poly(ethyleneoxy)ethanol, 1 mM Na3VO4, 48.8 mM NaF, 8.2 mM Na4P2O7, and 0.26 M saccharose | |||
| 6x Laemmli buffer | 1.2 g SDS, 6 mg bromophenol blue, 4.7 mL glycerol, 1.2 mL Tris base 0.5 M pH 6.8, 845 µL 2- mercaptoethanol, and 2.1 mL H2O | |||
| Running buffer | 25 mM Tris base, 192 mM glycine, 1% SDS | |||
| Transfer buffer | 25 mM Tris-base, 192 mM glycine, 20% methanol | |||
Table 1: Solutions used in this protocol.
Supplementary Figure S1: Subcutaneous adipose tissue digested with collagenase. Once the adipose tissue is removed, it is cut into small pieces with scissors to start the digestion process (A), then it is incubated for 30 min with collagenase type 1 at 37 °C, 150 rpm (B). Please click here to download this File.
Supplementary Figure S2: Scheme of the adipocyte precursor cell isolation process. Dissect the inguinal subcutaneous adipose tissue and digest the samples with collagenase. Eliminate mature adipocytes by centrifugation and wash the pellet to remove excess fat. Lyse the red blood cells and block to reduce nonspecific binding. Label the endothelial cells and macrophages with antibodies coupled to magnetic particles. Perform the magnetic separation of cells using the negative separation strategy with a magnetic cell separator. Seed APCs (unlabeled cells) in plates covered with basement membrane matrix. Change the medium every 48 h until the cells reach 80% confluence. Abbreviations: APCs = adipocyte precursor cells; BSA = bovine serum albumin; FBS = fetal bovine serum. Created with Biorender.com. Please click here to download this File.
Supplementary Figure S3: Treatment with TNF-α to induce insulin resistance does not alter the viability of adipocytes. The viability of mature adipocytes was measured by the MTT assay after treatment with 4 ng/mL of TNF-α for 48 h. Data are mean ± SEM. Abbreviations: TNF-α = tumor necrosis factor-α; Ctrl = control. Please click here to download this File.
Supplementary Figure S4: TNF-α treatment in adipocytes decreases the expression of insulin sensitivity markers. The mRNA expression of Insr, Irs, Glut4, and Adipoq was measured by real-time PCR after treatment with 4 ng/mL of TNF-α for 48 h. Data are mean ± SEM; *, p < .05 versus control. Abbreviations: TNF-α = tumor necrosis factor-α; Ctrl = control. Please click here to download this File.
