1. Washing the coverslips
NOTE: Wash (24 mm x 60 mm, #1.5) coverslips according to Smith et al., 201328.
- Arrange coverslips in a plastic slide mailer container.
- Submerge coverslips sequentially in the following solutions and sonicate for 30-60 min, rinsing with ddH2O 10 times in between each solution: ddH2O with one drop of dish soap; 0.1 M KOH. Store coverslips in 100% ethanol for up to 6 months.
NOTE: Do not touch glass surfaces with ungloved fingers. Use forceps instead.
2. Coating cleaned (24 mm x 60 mm, #1.5) coverslips with mPEG- and biotin-PEG-silane
NOTE: This protocol specifically uses a biotin-streptavidin system to position actin and microtubules within the TIRF imaging plane. Other coatings and systems may be used (e.g., antibodies, poly-L-lysine, NEM myosin, etc.).
- Thaw aliquots of PEG-silane and biotin-PEG-silane powders.
- Dissolve PEG powders in 80% ethanol (pH 2.0) to generate coating stock solutions of 10 mg/mL mPEG-silane and 2-4 mg/mL biotin-PEG-silane, just before the use.
NOTE: PEG powders often appear dissolved but may not be at the microscopic level. Proper resuspension takes ~1-2 min with constant pipetting. Users are encouraged to pipette an additional 10 times following the appearance of powder dissolution.
CAUTION: Wear gloves to protect skin from concentrated HCl when making 80% ethanol (pH 2.0). - Remove the clean (24 mm x 60 mm, #1.5) coverslip from ethanol storage using forceps. Dry with nitrogen gas and store in a clean Petri dish.
- Coat coverslips with 100 µL of coating solution: a mixture of 2 mg/mL mPEG-silane (MW 2,000) and 0.04 mg/mL biotin-PEG-silane (MW 3,400) in 80% ethanol (pH 2.0).
NOTE: For sparse coating (recommended) use 2 mg/mL mPEG-silane and 0.04 mg/mL biotin-PEG-silane. For dense coating use 2 mg/mL mPEG-silane, 4 mg/mL biotin-PEG-silane. - Incubate coverslips at 70 °C for at least 18 h or until use.
NOTE: Coated coverslips degrade if stored at 70 °C for more than 2 weeks.
3. Assembling imaging flow chambers
- Cut 12 strips of double-backed double-sided tape to a length of 24 mm. Remove one side of the tape backing and fix pieces of tape adjacent to the six grooves present on a clean imaging chamber.
NOTE: Tape must be flat for proper assembly, otherwise imaging chambers will leak. Carefully remove the tape backing to avoid bumps. Sliding taped chambers on a clean surface to smooth tape-chamber contacts is recommended. - Remove the second piece of tape backing to expose the sticky side of the tape along each chamber groove. Place chamber tape side up on a clean surface.
- Mix epoxy resin and hardener solutions 1:1 (or according to manufacturer's instructions) in a small weigh boat.
- Use a P1000 tip to place a drop of mixed epoxy between the tape strips at the end of each imaging chamber groove (red arrow; Figure 1A). Place chamber tape/epoxy side up on a clean surface.
- Remove a coated coverslip from 70 °C incubator. Rinse coated and uncoated surfaces of coverslips with ddH2O six times, dry with filtered nitrogen gas, and then affix to the imaging chamber with the coverslip coating side toward the tape.
- Use a P200 or P1000 pipette tip to apply pressure on the tape-glass interface to ensure a good seal between the tape and the coverslip.
NOTE: With a proper seal, double-sided tape becomes translucent. Imaging chambers lacking sufficient tape-chamber contacts will leak. - Incubate assembled chambers at room temperature for at least 5-10 min to allow the epoxy to fully seal chamber wells before use. Perfusion chambers expire within 12-18 h of assembly.
NOTE: Depending on tape placement and the thickness of double-sided tape used, the assembled chamber will have a final volume of 20-50 µL.
4. Conditioning of perfusion chambers
- Use a perfusion pump (rate set to 500 µL/min) to sequentially exchange conditioning solutions in perfusion chamber as follows:
- Flow 50 µL of 1% BSA to prime the imaging chamber. Remove excess buffer from Luer lock fitting reservoir.
- Flow 50 µL of 0.005 mg/mL streptavidin. Incubate for 1-2 min at room temperature. Remove excess buffer from reservoir.
- Flow 50 µL of 1% BSA to block nonspecific binding. Incubate for 10-30 s. Remove excess buffer from reservoir.
- Flow 50 µL of warm (37 °C) 1x TIRF buffer (1x BRB80, 50 mM KCl, 10 mM DTT, 40 mM glucose, 0.25% (v/v) methylcellulose (4,000 cp)).
NOTE: Do not remove excess buffer from the reservoir. This prevents the chamber from drying out, which can introduce air bubbles into the system. - Optional: Flow 50 µL of stabilized29 and 50% biotinylated microtubule seeds diluted in 1x TIRF buffer.
NOTE: The proper dilution must be empirically determined and contain batch to batch variability. Protocols from27,29 are recommended as starting points. A dilution yielding 10-30 seeds per field of view works well with this setup.
5. Microscope preparation
NOTE: Biochemical reactions containing dynamic actin filaments and microtubules are visualized/performed using an inverted Total Internal Reflection Fluorescence (TIRF) microscope equipped with 120-150 mW solid-state lasers, a temperature corrected 63x oil immersion TIRF objective, and an EMCCD camera. Proteins in this example are visualized at the following wavelengths: 488 nm (microtubules) and 647 nm (actin).
- Set the stage/objective heater device to maintain 35-37 °C at least 30 min prior to imaging the first biochemical reaction.
- Set the image acquisition parameters as follows:
- Set acquisition interval to every 5 s for 15-20 min.
- Set 488 and 647 laser exposures to 50-100 ms at 5%-10% power. Set appropriate TIRF angle for microscope.
NOTE: Regardless of microscope setup, the simplest way to set the laser power, exposure, and TIRF angle is to make adjustments on images of either polymer alone (see 5.2.2.1 and 5.2.2.2, below). Users are strongly encouraged to use the lowest laser power and exposure settings that still permit detection.- Adjust the polymerization reaction (Figure 1C) to initiate actin filament assembly and acquire images at 647 nm. Make appropriate adjustments.
- Adjust the polymerization reaction in a second conditioned perfusion well to initiate microtubule assembly (Figure 1C) and visualize at 488 nm. Make appropriate adjustments.
6. Preparation of protein reaction mixes
- Prepare stock solution of fluorescently labelled tubulin.
- Determine the concentration of homemade unlabeled tubulin via spectrophotometry at Abs280, as follows:
- Blank spectrophotometer with 1xBRB80 lacking GTP.
- Calculate the concentration of tubulin using the determined extinction coefficient of 115,000 M-1 cm-1 and the following formula:
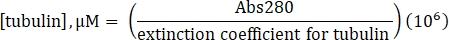
- Resuspend commercially-made lyophilized lysine-labeled 488-tubulin to 10 µM (1 mg/mL; 100% label) with 20 µL of 1x BRB80 lacking GTP.
- Thaw a 7.2 µL aliquot of 100 µM unlabeled recycled tubulin29 on ice.
NOTE: Recycled tubulin is critical for successful microtubule assembly in vitro because it removes polymerization-incompetent dimers formed in frozen protein stocks29,30. - Combine 3 µL of 10 µM 488-tubulin with the 7.2 µL aliquot of 100 µM unlabeled tubulin, no more than 15 min before use.
- Determine the concentration of homemade unlabeled tubulin via spectrophotometry at Abs280, as follows:
- Prepare the stock solution of fluorescently labeled actin.
- For homemade proteins, determine the concentration and percent label of actin via spectrophotometry Abs290 and Abs650, as follows:
- Blank spectrophotometer with G-buffer.
- Calculate the concentration of unlabeled actin using the determined extinction coefficient of 25,974 M-1 cm-1 and the following formula:
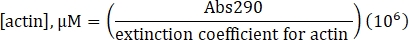
- Calculate the concentration of lysine labeled Alexa-647-actin using the extinction coefficient of unlabeled actin, the fluor correction factor of 0.03, and the following formula:
[Alexa-647 actin], µM = (Abs290 -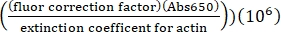
- Calculate the percent label of Alexa-647-actin using the determined ε for Alexa-647 of 239,000 M-1 cm-1, as follows:
% label of Alexa-647-actin = (Abs290 – (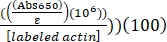
- Thaw one 2 µL aliquot of 3 µM 100% labeled biotin-actin (labeled on lysine residues). Dilute 10-fold by adding 18 µL of G-buffer.
- Combine 3 µL of diluted biotinylated actin, appropriate volumes of unlabeled and labeled actin (above) such that the final mix will be 12.5 µM total actin with 10%-30% fluorescent label.
NOTE: Greater than 30% percent fluorescent actin monomers (final) can compromise imaging resolution as filaments become difficult to discern from the background.
- For homemade proteins, determine the concentration and percent label of actin via spectrophotometry Abs290 and Abs650, as follows:
- Prepare reaction mixes (Figure 1C).
- Prepare cytoskeleton mix (Tube A) by combining 2 µL of the 12.5 µM actin mix stock (6.2.3) with the tubulin stock mix (6.1.4), no more than 15 min prior to imaging. Store on ice until use.
- Prepare protein reaction mix (Tube B) by combining all other experimental components and proteins, including: 2x TIRF buffer, anti-bleach, nucleotides, buffers, and accessory proteins. An example is shown in Figure 1C.
NOTE: The final dilution results in a 1x TIRF buffer that contains ATP, GTP, and ionic strength within the estimated physiological range.
- Incubate Tube A and Tube B separately at 37 °C for 30-60 s. To initiate reaction, mix and add the contents of Tube B to Tube A (below).
7. Image actin and microtubule dynamics
- Condition perfusion well (Figure 1B; step 4, above).
- Initiate actin and microtubule assembly simultaneously by adding the contents of Tube B (reaction mix) to Tube A (cytoskeleton mix) (Figure 1C).
- Flow 50 µL of reaction containing 1x TIRF buffer supplemented with 15 µM free tubulin, 1 mM GTP, and 0.5 µM actin monomers and appropriate volumes of buffer controls.
- Record time-lapse movie using microscope software to acquire every 5 s for 15-20 min.
NOTE: Initiation of actin and microtubule dynamics occurs within 2-5 min (Figure 2). Longer delays indicate problems with temperature control or concentration-related problems of proteins in the reaction mix. - Condition a new perfusion well (step 4) and replace buffer volume with regulatory protein(s) of interest (i.e., Tau) and buffer controls (Figure 1C). Acquire as outlined in step 7 (above) to assess regulatory proteins for emergent actin-microtubule functions.
8. Process and analyze images using FIJI software31
- Open saved TIRF movies and view as a composite.
- Analyze microtubule dynamics (Figure 3A), as follows:
- Generate a time-based maximum Z-projection from the image stacks menu.
- Synchronize Z-projection window with the original TIRF movie from the Analyze> Tools> Synchronize Windows menu.
- Draw a line using the straight-line tool along a microtubule of interest on the time projected image.
- Open the region of interest (ROI) manager from the analyze menu (Analyze> Tools> ROI Manager).
- Save individual microtubule locations by pressing "t". Repeat for all microtubules of interest.
- Plot kymographs of selected lines using "/" or run the multi-kymo macro that generates a video and kymograph for every microtubule in the ROI manager31.
- Add both length (µm) and time (min) scale bars to kymographs from the Analyze> Tools> Scale bars menu.
- Measure microtubule growth speeds from kymograph slopes (Figure 3A, 1-2; slope of black lines).
- Count dynamic microtubule events (catastrophe or regrowth) from the generated kymograph or using available analysis macros5,8,18,25. Red dotted lines in Figure 3A, 1-2 represent catastrophe/rapid disassembly events.
- Analyze actin dynamics (Figure 3B), as follows:
- Measure actin nucleation, as follows:
- Count the number of actin filaments present in the field of view 100 s after the initiation of the reaction and express by the area (filaments per µm2).
- Record and save data in ROI manager, as in step 8.2.3.1, above.
- Measure actin filament elongation rates (Figure 3B), as follows:
- Draw a line along an actin filament of interest using the segmented-line tool.
- Add line to ROI manager as in step 8.2.3.1, above.
- Repeat following the line (adding each measurement to ROI manager) for at least four movie frames.
NOTE: Measuring seven to eight consecutive frames is recommended, however some conditions slow actin polymerization below the detectable limit that can be resolved by objective/microscope setups. In such a case, measurements can be made at regular intervals over non-consecutive frames, (e.g., every five frames). - Plot measured length values over elapsed time. The slope of the generated line is the actin elongation rate in microns/s.
- Convey final calculated rates as subunits s-1 µM-1 using a correction factor of 370 subunits to account for the number of actin monomers in a micron of filament32.
- Measure actin nucleation, as follows:
- Perform correlative analysis for regions of parallel actin-microtubule association (Figure 3C), as follows:
- Draw a line along a microtubule of interest at a specific time point (i.e., 300 s after reaction initiation), using the straight-line tool.
- Add line to ROI manager as in step 8.2.3.1, above.
- Plot the fluorescence intensity along the line in each channel.
- Select each channel with the image slider and plot the intensities along the line using "command k".
- Save or export values by clicking the "list" button in the output window.
- Express actin-microtubule coupling events as a ratio (actin overlapping with microtubules) from individual events or as a count of events in a given field of view at a consistent timepoint (Figure 3C).
- Alternative: Use software to determine the percent overlap of both channels5,12.
- Draw a line along a microtubule of interest at a specific time point (i.e., 300 s after reaction initiation), using the straight-line tool.
With the conditions described above (Figure 1), actin and microtubule polymers should be visible (and dynamic) within 2 min of image acquisition (Figure 2). As with any biochemistry-based protocol, optimization may be required for different regulatory proteins or batches of protein. For these reasons, the TIRF angle and image exposures are set first with reactions containing each individual polymer. This confirms that stored proteins are functional and enough labeled protein is present for detection. While not always necessary (and not performed here), post-processing of movies (i.e., background subtraction, averaging, or Fourier transformations) can be used to enhance the image contrast (particularly of microtubules)5,25,33. The direct visualization of single actin filaments and microtubules afforded by this assay supports the quantitative determination of several dynamic measures for either cytoskeleton component alone or together, including polymerization parameters (i.e., nucleation or elongation rate), disassembly parameters (i.e., shrinkage rates or catastrophe events), and polymer coalignment/overlap (Figure 3). Further, these measures can be used as a starting point to decipher the binding or influence of regulatory ligands like Tau (Figure 3). Many measurements of single actin filaments or microtubules can be made from one TIRF movie. However, due to variations in coverslip coating, pipetting, and other factors, reliable measurements should also include multiple technical replicate reactions/movies.
Many facets of microtubule dynamics can be determined from example kymographs including the rate of microtubule elongation, as well as the frequency of catastrophe and rescue events (Figure 3A). Using kymographs to measure actin dynamics in this system is not as straightforward because actin filaments are more convoluted than microtubules. As a consequence, parameters of actin filament dynamics are measured by hand, which is time consuming and labor intensive. Nucleation counts are measured as the number of actin filaments present at a consistent timepoint for all conditions. These counts vary widely across TIRF imaging fields, but can be used with many replicates or to supplement observations from other polymerization assays. Nucleation counts may also be used for microtubules if trial conditions lack stabilized microtubule seeds. Actin filament elongation rates are measured as the length of filament over time from at least four movie frames. Rate values are conveyed per micromolar actin with a correction factor of 370 subunits to account for the number of actin monomers in a micron of filament (Figure 3B)32. Measurements to define the coordinated behaviors between actin and microtubules are less well defined. However, correlative analyses have been applied to measure the coincidence of both polymers including line scans (Figure 3C) or overlap software5,11,34.
Data Availability:
All datasets associated with this work have been deposited in Zenodo and are available with reasonable request at: 10.5281/zenodo.6368327.
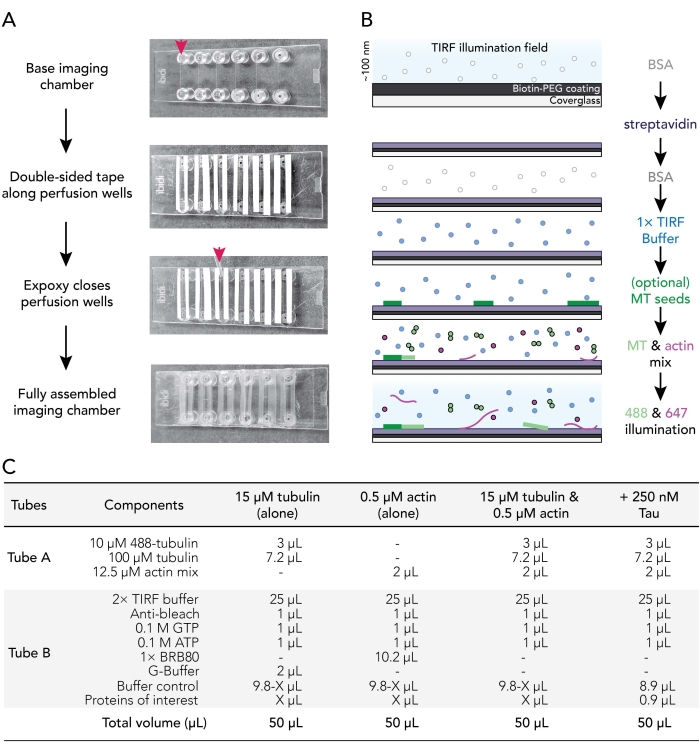
Figure 1. Experimental schematics: flow chamber assembly to image acquisition. (A) Imaging chamber assembly. Top to bottom: IBIDI imaging chambers are taped along perfusion wells (denoted by arrow); the second (white) layer of tape backing (left on in the image shown to better orient users) is removed and Epoxy is applied at the edge of the perfusion chamber (arrow). Note: To more easily orient users where to place the epoxy, the white backing was left on in this image. The cleaned and coated coverslip is attached to the imaging chamber with the coating side facing the inside of the perfusion well. (B) Flow-chart illustrating the steps for conditioning imaging chambers for biotin-streptavidin linkages. (C) Examples of reactions used to acquire TIRF movies of dynamic microtubules and actin filaments. Please click here to view a larger version of this figure.
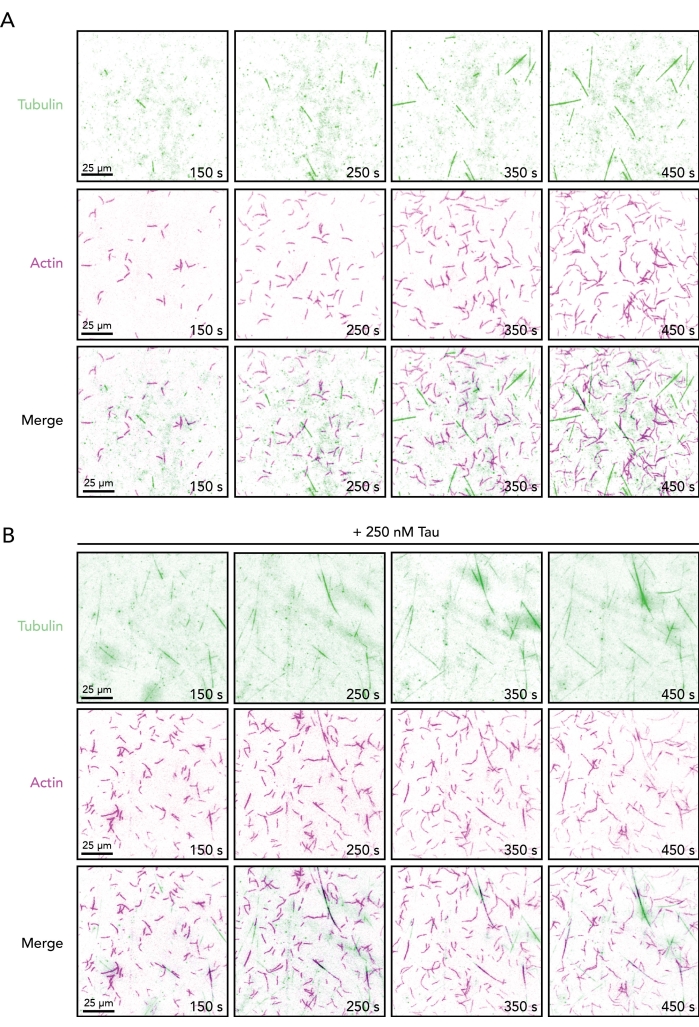
Figure 2. Image sequences of growing actin filaments and microtubules in the absence or presence of Tau. Time-lapse image montage from TIRF assays containing 0.5 µM actin (10% Alexa-647-actin and 0.09% biotin-actin labeled) and 15 µM free tubulin (4% HiLyte-488 labeled) in the absence (A) or presence (B) of 250 nM Tau. Time elapsed from reaction initiation (mixing Tube A and Tube B) is shown. Scale bars, 25 µm. Please click here to view a larger version of this figure.
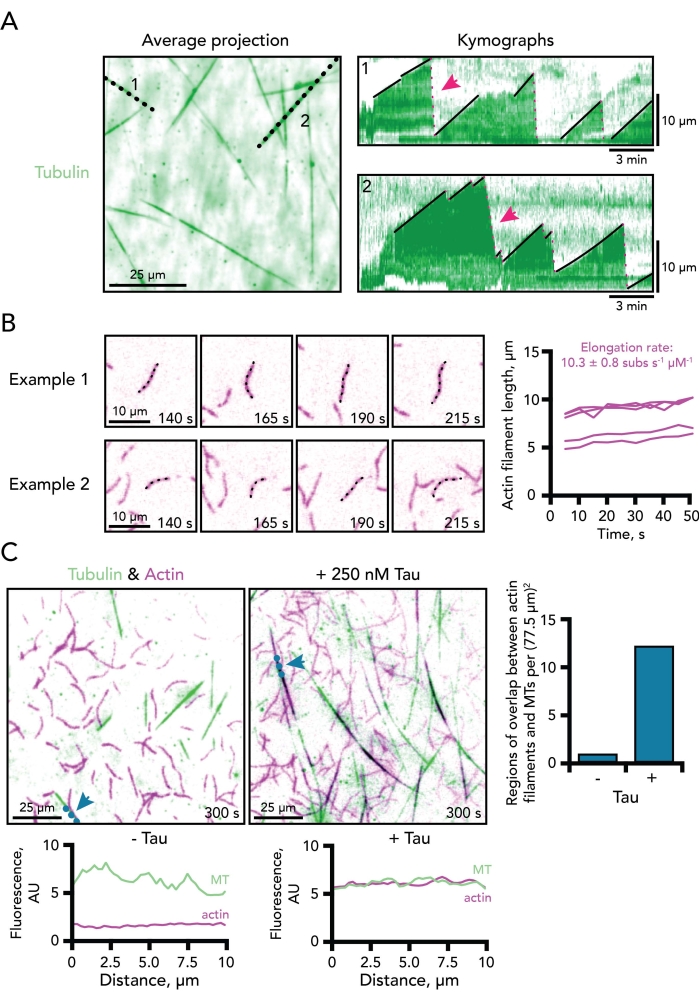
Figure 3. Example measurements of microtubules and actin filament dynamics. (A) Average time projection of the tubulin channel efficiently visualizes total microtubule lengths for the line scans used to generate kymograph plots. Black dotted lines correspond to the two example kymographs of dynamic microtubules shown on the right. The growth (solid black lines) and disassembly phases (doted pink lines; two denoted with pink arrows) of microtubules are shown on each kymograph. Time scale bar, 3 min. Length scale bar, 10 µm. Reaction contains 0.5 µM actin (10% 647-label) and 15 µM free tubulin (4% 488-HiLyte label). Only the tubulin channel is shown.(B) Two example time-lapse image montages depicting single-actin filaments actively polymerizing. Elongation rates are calculated as the slope of plots of the length of actin filaments over time per micromolar actin. Thus, a correction factor of two must be applied to 0.5 µM actin reactions for comparison for rates typically determined at the 1 µM actin concentration. Examples from five filaments are shown to the right. Scale bars, 10 µm. Reaction contains 0.5 µM actin (10% 647-label) and 15 µM free tubulin (4% 488-HiLyte label). Only the actin channel is shown. (C) TIRF images of dynamic microtubules (MT) (green) and actin filaments (purple) polymerizing in the absence (left) or presence of 250 nM Tau (middle). Blue dotted lines and arrows mark where a line was drawn for the line scan plots corresponding to each condition (below each image). Overlap between microtubules and actin regions (shown as black) can be scored at a set time point per area (right). Scale bars, 25 µm. Reactions contain 0.5 µM actin (10% 647-label) and 15 µM free tubulin (4% 488-HiLyte label) with or without 250 nM Tau. Please click here to view a larger version of this figure.
