ヒト造血幹細胞および前駆細胞における発癌性ヘテロ接合性機能獲得変異の改変
Summary
造血幹細胞生物学および造血悪性腫瘍をよりよく研究するためには、造血幹細胞および前駆細胞(HSPC)の体細胞変異を忠実にモデル化するための新しい戦略が必要です。ここでは、CRISPR/Cas9とデュアルrAAVドナー形質導入の使用を組み合わせることにより、HSPCにおけるヘテロ接合機能獲得変異をモデル化するためのプロトコルについて説明します。
Abstract
その生涯を通じて、造血幹および前駆細胞(HSPC)は体細胞変異を獲得します。これらの変異のいくつかは、増殖や分化などのHSPC機能特性を変化させ、それによって血液悪性腫瘍の発症を促進します。HSPCの効率的かつ正確な遺伝子操作は、再発性の体細胞突然変異の機能的影響をモデル化し、特徴付け、よりよく理解するために必要です。突然変異は遺伝子に有害な影響を及ぼし、機能喪失(LOF)をもたらすか、まったく対照的に、機能を強化したり、機能獲得(GOF)と呼ばれる特定の遺伝子の新しい特性を引き起こしたりする可能性があります。LOF変異とは対照的に、GOF変異はほぼ独占的にヘテロ接合方式で発生します。現在のゲノム編集プロトコルでは、個々の対立遺伝子の選択的標的化はできず、ヘテロ接合型のGOF変異をモデル化する能力を妨げています。ここでは、効率的なDNAドナーテンプレート導入のために、CRISPR/Cas9を介した相同性指向修復と組換えAAV6技術を組み合わせることにより、ヒトHSPCにおけるヘテロ接合型GOFホットスポット変異を設計する方法に関する詳細なプロトコルを提供します。重要なことに、この戦略は、ヘテロ接合的に編集されたHSPCの追跡と精製を可能にするために、デュアル蛍光レポーターシステムを利用しています。この戦略は、GOF変異がHSPCの機能にどのように影響するか、および血液悪性腫瘍への進行を正確に調査するために使用できます。
Introduction
クラスター化された規則的に間隔を空けた短い回文反復(CRISPR)/ Cas9技術の開発により、新しい非常に強力な機器が科学者のツールキットに追加されました。この技術はゲノムの精密なエンジニアリングを可能にし、研究目的(Hsu et al.1でレビュー)だけでなく、最近では臨床現場への変換にも成功していることが証明されています2,3,4。CRISPR/Cas9編集戦略は、Cas9タンパク質とシングルガイドRNA(sgRNA)の活性に依存しています5,6,7。宿主細胞では、Cas9タンパク質はsgRNA配列に相補的なDNAの特定の部位に誘導され、DNA二本鎖切断(DSB)を導入します。DSBが生成されると、非相同末端結合(NHEJ)と相同性指向修復(HDR)の2つの主要な競合する修復メカニズムが発生する可能性があります。NHEJはエラーが発生しやすく、主に挿入と欠失(インデル)につながる修復メカニズムですが、姉妹染色分体を修復テンプレートとして使用するHDRは非常に正確ですが、細胞周期のSまたはG2期に限定されます8。ゲノム工学では、Cas9誘導性DSBの両DNA末端と同一の相同性アームに隣接するドナーテンプレートを提供することにより、HDRをDNAの標的修飾に利用できます(図1)。HDR に使用されるドナーテンプレートのタイプは、編集効率に大きな影響を与える可能性があります。ヒトHSPCにおける遺伝子工学に関して、アデノ随伴ウイルス血清型6(AAV6)は、一本鎖DNA鋳型の送達のための優れた媒体であることが最近記載されている9、10。
CRISPR/Cas9ゲノム工学は、有害な変異を修正するために治療的に使用することができます11が、癌の発生をモデル化するためにDNAに病原性突然変異を導入するために利用することもできます12。白血病などの血液がんは、健康なHSPCにおける体細胞変異の順次獲得を介して発症します13,14。初期の遺伝的事象はクローン増殖の利点をもたらし、不確定な可能性(CHIP)のクローン造血をもたらします15,16。突然変異のさらなる獲得は、最終的に白血病の形質転換と病気の発症につながります。体細胞変異は、自己複製、生存、増殖、および分化を制御する遺伝子に見られます17。
ゲノムエンジニアリングを介して個々の突然変異を健康なHSPCに導入することで、この段階的な白血病発生プロセスを正確にモデル化することができます。急性骨髄性白血病(AML)18,19などの骨髄性新生物に見られる再発性変異の数が限られているため、この疾患はゲノム工学ツールを使用して再現するのに特に適しています。
体細胞変異は、一方の対立遺伝子(単対立遺伝子/ヘテロ接合型変異)または両方の対立遺伝子(双対立遺伝子/ホモ接合型変異)にのみ出現し、遺伝子の機能に大きな影響を与える可能性があり、機能喪失(LOF)または機能獲得(GOF)を引き起こす可能性があります。LOF変異は、遺伝子のLOFの低下(一方の対立遺伝子が影響を受ける場合)または完全な(両方の対立遺伝子が影響を受ける場合)をもたらすが、GOF変異は、遺伝子の活性化または新規機能の増加をもたらす。GOF変異は、典型的にはヘテロ接合型20である。
重要なことに、接合性(ヘテロ接合対ホモ接合)は、突然変異を忠実にモデル化する試みに大きな影響を及ぼします。したがって、遺伝子の1つの対立遺伝子のみの標的操作は、ヘテロ接合ホットスポットGOF変異を設計するために必要です。エラーが発生しやすいNHEJは、さまざまな長さのインデル21につながり、さまざまな予測不可能な生物学的結果につながる可能性があります。しかし、NHEJはDSBの導入後に細胞によって採用された主要な修復プログラムであるため、HSPCを操作するために現在使用されているほとんどのCRISPR/Cas9プラットフォームでは、遺伝的転帰を正確に予測することができません22,23。対照的に、CRISPR/Cas9を介した二本鎖切断(DSB)の導入と、HDRを介したゲノムエンジニアリングのための組換えアデノ随伴ウイルス(rAAV)ベクターベースのDNAドナーテンプレートの使用を組み合わせることで、ヒトHSPCにおける変異の対立遺伝子特異的挿入が可能になります11,24。変異体と野生型(WT)配列の同時統合を、個々の対立遺伝子上の異なる蛍光レポーターと組み合わせて実行して、ヘテロ接合遺伝子型を選択できます(図2)。この戦略は、HSPCの機能、疾患の開始、および進行に対する再発性、白血病性、ヘテロ接合性のGOFホットスポット変異の影響を正確に特徴付けるための強力なツールとして活用できます。
この記事では、初代ヒトHSPCにおける反復変異ヘテロ接合型GOF変異の効率的なエンジニアリングのための詳細なプロトコルを提供します。この戦略は、CRISPR/Cas9とデュアルAAV6形質導入の使用を組み合わせて、ヘテロ接合型GOF変異の将来の生成のためのWTおよび変異DNAドナーテンプレートを提供します。一例として、カルレティキュリン(CALR)遺伝子における再発性1型変異(52bp欠失)の工学が図25に示される。CALRのエクソン9におけるヘテロ接合型GOF変異は、本態性血小板血症(ET)や原発性骨髄線維症(PMF)などの骨髄増殖性疾患に再発見られます26。CALRは小胞体常在タンパク質であり、主に新規合成タンパク質の折り畳み過程における品質管理機能を有する。その構造は、タンパク質のシャペロン機能に関与するアミノ(N)末端ドメインとプロリンリッチPドメイン、およびカルシウム貯蔵と調節に関与するCドメインの3つの主要なドメインに分けることができます27,28。CALR変異は+1フレームシフトを引き起こし、新規の伸長したC末端の転写と小胞体(ER)保持シグナル(KDEL)の喪失につながります。変異型CALRはトロンボポエチン(TPO)受容体に結合することが示されており、それによって増殖の増加を伴うTPO非依存性シグナル伝達をもたらす29。
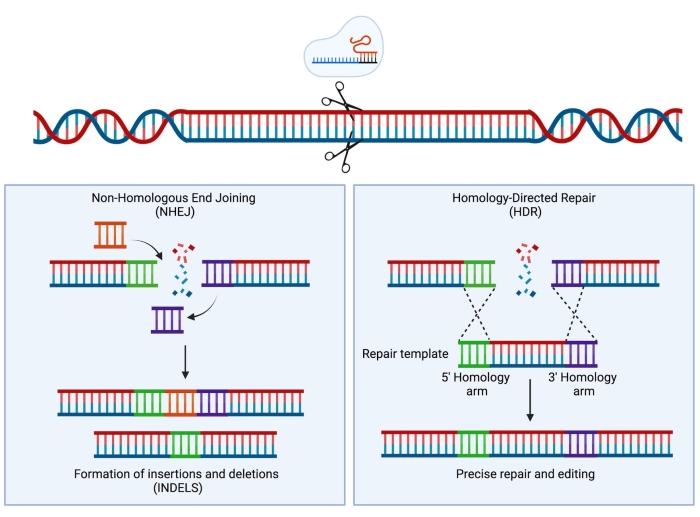
図1:NHEJとHDRの修復。 DNAに二本鎖切断を導入した後のNHEJおよびHDR修復メカニズムの簡略化された概略図。 この図の拡大版を表示するには、ここをクリックしてください。
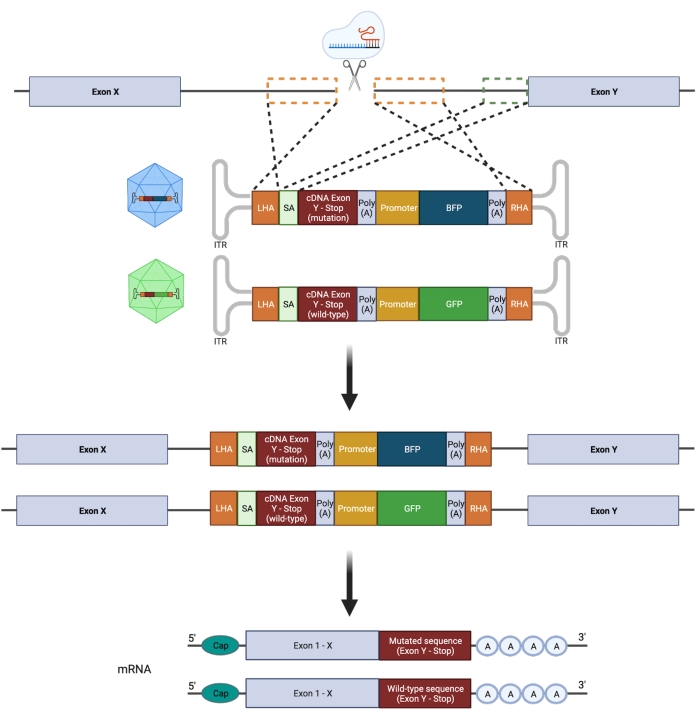
図2:双対立遺伝子HDR編集戦略の概略図。 ドナーテンプレートの標的対立遺伝子への統合とそれに続く機能的mRNAへの翻訳を示す概略図。オレンジ色の点線のボックスは、左相同性アーム(LHA)および右相同性アーム(RHA)に対応する領域を示す。HA の理想的なサイズは、それぞれ 400 bp です。緑色の点線のボックスは、SA配列に対応する領域を表す。SAのサイズは150 bpです。 この図の拡大版を表示するには、ここをクリックしてください。
Protocol
Representative Results
Discussion
ヒト初代HSPCの効率的かつ正確な遺伝子操作は、正常な造血、そして最も重要なことに、造血細胞の白血病形質転換に影響を与えるプロセスを探求し理解する絶好の機会を表しています。
このプロトコルでは、再発性ヘテロ接合型GOF変異を発現するようにヒトHSPCを設計するための効率的な戦略が説明されました。この手順では、CRISPR/Cas9テクノロジーとrAAV6ベクターをDNAテンプレートのドナーとして利用して、WTおよび変異DNA配列を内因性遺伝子座に正確に挿入しました。改変されたcDNA(WTおよび変異体)を分離された蛍光レポータータンパク質と結合させることで、決定的なヘテロ接合状態にある細胞の濃縮と追跡が可能になります。
この戦略は、頻繁に使用されるレンチウイルス(LV)ベースの方法と比較していくつかの利点を提示します。主な利点の1つは、CRISPR/Cas9ベースのシステムにより、内因性遺伝子座の正確な編集が可能になり、内因性プロモーターと調節要素が保存されることです。これは、細胞内で編集された遺伝子の発現の均一性をもたらし、LVベースの方法が使用される場合には達成不可能な目標である。LVベクターによる遺伝子導入は、転写活性部位34を優先して遺伝子の半ランダム組込みをもたらす。これは、移入された遺伝子の過剰発現と編集された細胞間の不均一性に変換され、最終的には突然変異と遺伝子相互作用の役割を調査および分析することが困難になります。第二の利点は、記載されたシステムが、部位特異的編集システムであることであり、挿入突然変異誘発のリスクを排除することである35。
二重蛍光レポーター戦略は、両方の対立遺伝子で正常に編集された細胞の正確な濃縮と追跡を可能にし、一方の対立遺伝子がWT cDNAを統合し、もう一方の対立遺伝子が変異したcDNA配列を統合します。単一のレポーターのみを発現する細胞は、同じ蛍光レポーターを有するHDRテンプレートの単対立遺伝子統合または双対立遺伝子統合のいずれかを表す。どちらのシナリオも、単一細胞由来のクローンを作製し、個別に分析した場合にのみ正確に区別できます。しかし、HSPCは in vitroでの増殖能力が限られており、長期間培養しておくと、より成熟した子孫に分化し始め、自己複製能力と生着能力を失います。これにより、所望のヘテロ接合変異を有する単一細胞クローンの選択および増殖が不可能になる。ヘテロ接合変異を有する細胞に対するフローサイトメトリーによる二重蛍光タンパク質戦略および濃縮の適用は、長時間の in vitro 培養によって誘発される問題を回避することを可能にする。
この特定の例では、ヘテロ接合性のCALR DEL/WT変異を有するHSPCの純粋な集団を得るために、HSPCを効率的に操作および分類できることが実証されました。
しかし、このシステムは、ヘテロ接合型フレームシフト変異のエンジニアリングに限定されず、ミスセンス変異やナンセンス変異を含む他の変異タイプを作成するために容易に採用することもできます。WTを含むAAVまたは異なる蛍光レポータータンパク質を有する変異配列の異なる組み合わせを適用することにより、このシステムは、ホモ接合型変異の導入(変異cDNAを有するが蛍光レポーターが異なる2つのrAAVによる同時形質導入)または変異の補正(WT cDNAを有するが蛍光レポーターが異なる2つのAAVによる同時形質導入)にも利用することができる。さらに、この戦略は発癌性GOF変異の導入に限定されないことに言及することが重要です。実際、記載されたプロトコルは、遺伝子ノックアウト、遺伝子置換36,37、導入遺伝子(すなわち、キメラ抗原受容体)の標的ノックイン38、さらには疾患の原因となる突然変異の補正11,39を含む複数の代替戦略に利用することができる。
CRISPR/Cas9およびAAV6を複数の蛍光レポーターと組み合わせる戦略は、T細胞、形質細胞様樹状細胞、人工多能性幹細胞、神経幹細胞、気道幹細胞など、他の多くの細胞タイプにも適用できることが示されています24,38,40,41,42,43,44.この戦略は、優れたキメラ抗原受容体(CAR)T細胞の産生のために実施することができる。例えば、CAR-T細胞におけるTGFBR2遺伝子のCRISPR/Cas9媒介ノックアウトが、抑制性TGF-βに富む腫瘍微小環境におけるそれらの機能を大幅に増加させることが最近発表された45。このようなアプローチは、CARを発現するようにT細胞を設計し、TGFBR2遺伝子の両方の対立遺伝子にCARを特異的に挿入する部位によってTGFBR2遺伝子をノックアウトするためのワンステッププロトコルを提供する可能性があります。さらに、このアプローチは、CARをT細胞受容体アルファ定数(TRAC)遺伝子46,47に組み込むことによってユニバーサルCAR T細胞を生成するのにも有用であり得る。
再現性を高め、細胞の効率的な編集を保証するには、いくつかの重要な考慮事項に注意する必要があります。細胞の編集を成功させるための主な重要なポイントは、(i)sgRNAの選択、(ii)HDRテンプレートの設計、および(iii)rAAV6の生産にあります。
良好な性能のsgRNAの選択は、HDRテンプレートを統合できる対立遺伝子の最大数を決定するため、非常に重要です。現在利用可能な多数のソフトウェアにより、候補sgRNAの検索が簡素化されました。関心領域を選択することにより、ソフトウェアは、所望の遺伝子座および望ましくない遺伝子座での編集の可能性をそれぞれ示すオンターゲットスコアおよびオフターゲットスコアを有する一連のsgRNAを提案することができる。これらのスコアは、以前に公開されたスコアリングモデル48、49に基づいて計算される。これは良好な性能のsgRNAを選択するための良い出発点ですが、 インシリコでの 予測性能はin vitroで効率的なsgRNAに必ずしも対応しないため、sgRNAの性能を確認する必要があります。したがって、最良のsgRNAを見つける可能性を高めるために、少なくとも3つのsgRNAを設計してテストすることを強くお勧めします。真の良好な性能を発揮するsgRNAが特定されたら、HDRテンプレートの設計を進めることをお勧めします。
HDRテンプレートを設計する際には、予防措置を考慮する必要があります。左右の相同性アーム(それぞれLHAとRHA)は、HAが短くなるとHDR周波数が低下する可能性があるため、それぞれsgRNAカット部位の上流と下流にそれぞれ400 bpにまたがる必要があります。HDR を介して 導入できるcDNAのサイズは、AAVのパッケージング機能(約4.7kb)に依存します。HDRテンプレート内で必須の多数の要素(LHA、RHA、SA、PolyA、プロモーター、および蛍光レポーター配列)により、変異またはWT cDNAの残りのスペースは限られています。これは、目的の突然変異が遺伝子の3’末端近くにある場合、または全体的に短いCDSを持つ遺伝子にある場合は問題になりません。しかしながら、突然変異が長いCDSを有する(AAVの残りのパッキングスペースを超える)遺伝子の転写開始側(TSS)の近くに位置する場合、この記載されたアプローチは実行不可能であり得る。この問題を回避するために、HDRテンプレートを2つのAAVに分割することに依存する戦略が最近Bakらによって開発されました。この戦略は、大きな遺伝子50の最終的なシームレスな統合を得るために、2つの別個のHDR媒介統合に依存する。
ウイルスの品質とその力価は、細胞のゲノム工学の成功を左右する追加の要因です。最適な収量を得るには、HEK293Tを培養液で維持しながら完全なコンフルエントに達しさせないことが重要です。理想的には、HEK293T細胞は、70%〜80%のコンフルエントに達したときに分割する必要があります。さらに、HEK293Tはウイルス産生能力を低下させる可能性があるため、長期間培養しないでください。新しいHEK293T細胞は、20継代後に解凍する必要があります。高いウイルス力価を得ることは、実験の効率と再現性を高めるために重要です。ウイルス力価が低いと、HSPCの形質導入に必要な大量のrAAV溶液に変換されます。原則として、核細胞に添加されるrAAV溶液は、HSPC保持培地の総体積の20%を超えてはなりません。AAV溶液の量が多いと、細胞死の増加、増殖の低下、形質導入効率の低下につながる可能性があります。したがって、ウイルス力価が低い場合は、ウイルスをさらに濃縮することをお勧めします。
要約すると、このプロトコルは、CRIPSR/Cas9およびrAAV6ドナーテンプレートと追加のデュアル蛍光レポーターを同時に使用することにより、ヒトHSPCを正確かつ効率的に操作するための再現可能なアプローチを提供します。このアプローチは、正常な造血幹細胞生物学と突然変異が白血病発生にもたらす寄与を研究する上で優れたツールであることが証明されています。
Disclosures
The authors have nothing to disclose.
Acknowledgements
この研究は、オーストリア科学基金(FWF、番号P32783およびI5021)からA.R.への助成金によってサポートされています。臨床研究助成金)、およびMEFOgraz。T.K.は白血病・リンパ腫協会の特別研究員です。
Materials
| 175 cm2 Cell Culture Flask, Vent Cap, TC-treated | Corning | 431080 | |
| 150 mm x 25 mm dishes | Corning | 430599 | |
| 293T | DSMZ | ACC 635 | https://www.dsmz.de/collection/catalogue/details/culture/ACC-635 |
| 4D Nucleofector Core Unit | Lonza | – | For nucleofection of human HSPCs use the DZ-100 program. |
| 4D Nucleofector X Unit | Lonza | – | |
| 500 ml Centrifuge Tube | Corning | 431123 | |
| 7-AAD | BD Biosciences | 559925 | |
| AAVpro Purification Kit | Takara | 6666 | |
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | 1081058 | |
| Avanti JXN-30 Ultracentrifuge | Beckman Coulter | – | |
| Benchling sgRNA design tool | Online tool for sgRNA design: http://www.benchling.com/crispr | ||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7906-100G | |
| C1000 Touch Thermal Cycler | Bio-Rad | – | |
| Chemically modified synthetic sgRNA | Synthego | Website: https://www.synthego.com/products/crispr-kits/synthetic-sgrna Sequence for the sgRNA targeting intron 7 of CALR: 5’-CGCCTGTAATCCTCGCCCAG-3’ An 80 nucleotide SpCas9 scaffold is added to the 20 nucleotide RNA sequence to complete the sgRNA. Chemical modifications of 2'-O-Methyl are added to the first and last 3 bases and 3' phosphorothioate bonds are added in the first 3 and last 2 bases. *Alternatively chemically modified synthetic sgRNAs can be acquired from IDT (https://eu.idtdna.com/site/order/oligoentry/index/crispr) and Trilink (https://www.trilinkbiotech.com/custom-oligos) | |
| CHOPCHOP sgRNA design tool | Online tool for sgRNA design: http://chopchop.cbu.uib.no | ||
| Costar 24-well Clear TC-treated Multiple Well Plates | Corning | 3526 | |
| CRISPick sgRNA design tool | Online tool for sgRNA design: https://portals.broadinstitute.org/gppx/crispick/public | ||
| CRISPOR sgRNA design tool | Online tool for sgRNA design: http://crispor.tefor.net | ||
| ddPCR 96-Well Plates | Bio-Rad | 12001925 | |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863024 | |
| DG8 Cartridges for QX200/QX100 Droplet Generator | Bio-Rad | 1864008 | |
| DG8 Gaskets for QX200/QX100 Droplet Generator | Bio-Rad | 1863009 | |
| DreamTaq Green PCR Master Mix (2X) | Thermo Scientific | K1081 | |
| Droplet Generation Oil for Probes | Bio-Rad | 1863005 | |
| Dulbecco’s Modified Eagle Medium (DMEM) with high glucose | Sigma-Aldrich | D6429-6X500ML | |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma-Aldrich | D8537-500ML | |
| FACSAria Fusion | BD Biosciences | – | |
| Falcon 5 mL Round Bottom | Corning | 352054 | |
| Fetal Bovine Serum (FBS) Good Forte (heat inactivated), 500 ml | Pan Biotech | P40-47500 | |
| FlowJo 10.8.0 | BD Biosciences | – | |
| GenAgarose L.E. | Inno-train | GX04090 | |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | |
| Gibson Assembly Master Mix | New England Biolabs Inc. (NEB) | E2611L | |
| HEK293T | |||
| HEPES solution | Sigma-Aldrich | H0887-100ML | |
| ICE | Synthego | https://ice.synthego.com | |
| IDT codon optimization tool | IDT | https://www.idtdna.com/pages/tools/codon-optimization-tool | |
| IDT sgRNA design tool | Online tool for sgRNA design: https://www.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM | ||
| LB Broth (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7658-1KG | |
| LB Broth with agar (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7533-1KG | |
| Midori Green Advance | Nippon Genetics | MG04 | |
| Monarch Plasmid Miniprep Kit | NEB | T1010L | |
| Monarch DNA Gel Extraction Kit | NEB | T1020L | |
| NEB 5-alpha Competent E. coli (High Efficiency) | NEB | C2987U | |
| Nuclease-Free Water, 5X100 ml | Ambion | AM9939 | |
| NucleoBond Xtra Midi | Macherey-Nagel | 740410 | |
| Opti-MEM, Reduced Serum Medium, 500 ml | Gibco | 31985070 | |
| P3 Primary Cell 4D-Nucleofetor X Kit L | Lonza | V4XP-3024 | The Lonza Primary P3 solution is supplied as a 2.25 mL P3 Primary Cell Nucleofector Solution and 0.5 mL Supplement 1. To reconstitute, add the Supplement 1 to the P3 Primary Cell Nucleofector Solution and mix. |
| pAAV-MCS2 | Addgene | 46954 | |
| PCR Plate Heat Seal Foil, pierceable | Bio-Rad | 1814040 | |
| pDGM6 | Addgene | 110660 | |
| Penicillin-Streptomycin (P/S) | Gibco | 15140122 | |
| Polyethylenimine (PEI) | Polysciences | 23966 | Add 50 mL of PBS 4.5 pH (made with HCl) to 50 mg of PEI in a tube. Dissolve by placing the tube in a 70°C water bath and vortexing every 10 minutes until the solution is dissolved. After the solution has reached RT, filter sterilze through a 0.22 μm filter, make 1120 μL aliquots, and store at -80°C. |
| Polystyrene Test Tube, with Snap Cap | |||
| Primers | Eurofins | – | Primers were ordered from Eurofins (eurofinsgenomics.eu) as unmodified salt free custom oligos. The primers were designed by using PRIMER-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) Primer 1 Fwd: AAGTGATCCGTTCGCCATGAC; Primer 2 Rev CALR WT specific: ACGTCCTCTTCCTCGTCCTC; Primer 2 Rev CALR DEL specific: CCAACCCTGGAGACACGCTTC |
| PrimeTime qPCR Primer Assay | IDT | – | PrimeTime qPCR Probe Assays (1 probe/2 primers) that can be ordered from IDT (https://eu.idtdna.com/site/order/qpcr/assayentry). Scale: Std – qPCR Assay 500 reactions; Primer 1 Forward (5'-3') : GGAACCCCTAGTGATGGAGTT; Primer 2 Reverse (5'-3'): CGGCCTCAGTGAGCGA; Probe (5'-3'): CACTCCCTCTCTGCGCGCTCG; 5' Dye/3' Quencher: FAM/ZEN/IBFQ; Primer to probe ratio: 3.6 |
| PX1 PCR Plate Sealer | Bio-Rad | 1814000 | |
| QuantaSoft Software | Bio-Rad | ||
| Quick Extract DNA Extraction Solution | Lucigen | QE0905T | |
| QX200 Droplet Generator | Bio-Rad | 1864002 | |
| QX200 Droplet Reader | Bio-Rad | ||
| Recombinant human Flt3-ligand | Peprotech | 300-19 | |
| Recombinant human IL-6 | Peprotech | 200-06 | |
| Recombinant Human SCF | Peprotech | 300-07 | |
| Recombinant Human TPO | Peprotech | 300-18 | |
| RPMI 1640 | Sigma-Aldrich | R8758-6X500ML | |
| SnapGene | Dotmatics | Molecular cloning software https://www.snapgene.com *Alternatively also Benchling (https://www.benchling.com) and Geneious (https://www.geneious.com) can be used. | |
| Soc outgrowth medium | NEB | B9020S | |
| Sodium-butyrate | Sigma-Aldrich | B5887-1G | |
| Stem Regenin 1 (SR1) | Biogems | 1224999 | |
| StemSpan SFEM II | STEMCELL Technologies | 9655 | |
| TAE Buffer (Tris-acetate-EDTA) 50X | Thermo Scientific | B49 | |
| TIDE | http://shinyapps.datacurators.nl/tide/ | ||
| Trypan blue 0.4% | Sigma-Aldrich | T8154-100ML | |
| TrypLE (with phenol red), 500 ml | Thermo Scientific | 16605-028 | |
| UltraPure 0.5: EDTA, pH 8.0, 100 ml | Thermo Scientific | 15575-038 | |
| UM171 | STEMCELL Technologies | 72914 | |
| Vector Builder codon optimization tool | Vector Builder | https://en.vectorbuilder.com/tool/codon-optimization.html |
References
- Hsu, P. D., Lander, E. S., Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157 (6), 1262-1278 (2014).
- Gillmore, J. D., et al. CRISPR-Cas9 in vivo gene editing transthyretin amyloidosis. New England Journal of Medicine. 385 (6), 493-502 (2021).
- Frangoul, H., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine. 384 (3), 252-260 (2021).
- Stadtmauer, E. A., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 367 (6481), (2020).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Heyer, W. D., Ehmsen, K. T., Liu, J. Regulation of homologous recombination in eukaryotes. Annual Review of Genetics. 44, 113-139 (2010).
- Veldwijk, M. R., et al. Pseudotyped recombinant adeno-associated viral vectors mediate efficient gene transfer into primary human CD34+ peripheral blood progenitor cells. Cytotherapy. 12 (1), 107-112 (2010).
- Song, L., et al. High-efficiency transduction of primary human hematopoietic stem cells and erythroid lineage-restricted expression by optimized AAV6 serotype vectors in vitro and in a murine xenograft model in vivo. PLoS One. 8 (3), 58757 (2013).
- Dever, D. P., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 539 (7629), 384-389 (2016).
- Drost, J., et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 521 (7550), 43-47 (2015).
- Jan, M., et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine. 4 (149), (2012).
- Corces-Zimmerman, M. R., Hong, W. J., Weissman, I. L., Medeiros, B. C., Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences of the United States of America. 111 (7), 2548-2553 (2014).
- Jaiswal, S., et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England Journal of Medicine. 377 (2), 111-121 (2017).
- Genovese, G., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. New England Journal of Medicine. 371 (26), 2477-2487 (2014).
- Papaemmanuil, E., et al. Genomic classification and prognosis in acute myeloid leukemia. New England Journal of Medicine. 374 (23), 2209-2221 (2016).
- Cancer Genone Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine. 368 (22), 2059-2074 (2013).
- Ball, M., List, A. F., Padron, E. When clinical heterogeneity exceeds genetic heterogeneity: thinking outside the genomic box in chronic myelomonocytic leukemia. Blood. 128 (20), 2381-2387 (2016).
- Varmus, H. E. The molecular genetics of cellular oncogenes. Annual Review of Genetics. 18, 553-612 (2003).
- Cox, D. B. T., Platt, R. J., Zhang, F. Therapeutic genome editing: Prospects and challenges. Nature Medicine. 21 (2), 121-131 (2015).
- Tothova, Z., et al. Multiplex CRISPR/Cas9-based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell. 21 (4), 547-555 (2017).
- Mandal, P. K., et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 15 (5), 643-652 (2014).
- Bak, R. O., Dever, D. P., Porteus, M. H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nature Protocols. 13 (2), 358-376 (2018).
- Foßelteder, J., et al. Human gene-engineered calreticulin mutant stem cells recapitulate MPN hallmarks and identify targetable vulnerabilities. Leukemia. , (2023).
- Nangalia, J., et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. New England Journal of Medicine. 369 (25), 2391-2405 (2013).
- Merlinsky, T. R., Levine, R. L., Pronier, E. Unfolding the role of calreticulin in myeloproliferative neoplasm pathogenesis. Clinical Cancer Research. 25 (10), 2956-2962 (2019).
- Belčič Mikič, T., Pajič, T., Zver, S., Sever, M. The contemporary approach to CALR-positive myeloproliferative neoplasms. International Journal of Molecular Sciences. 22 (7), 3371 (2021).
- How, J., Hobbs, G. S., Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood. 134 (25), 2242-2248 (2019).
- Grieger, J. C., Choi, V. W., Samulski, R. J. Production and characterization of adeno-associated viral vectors. Nature Protocols. 1 (3), 1412-1428 (2006).
- Zolotukhin, S., et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy. 6 (6), 973-985 (1999).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2011).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Bulcha, J. T., Wang, Y., Ma, H., Tai, P. W. L., Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduction and Targeted Therapy. 6 (1), 1-24 (2021).
- Montini, E., et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. The Journal of Clinical Investigation. 119 (4), 964-975 (2009).
- Vaidyanathan, S., et al. Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus. Molecular Therapy. 30 (1), 223-237 (2022).
- Cromer, M. K., et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nature Medicine. 27 (4), 677-687 (2021).
- Wiebking, V., et al. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing αβ T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica. 106 (3), 847-858 (2021).
- Wilkinson, A. C., et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nature Communications. 12 (1), 1-9 (2021).
- Dever, D. P., et al. CRISPR/Cas9 genome engineering in engraftable human brain-derived neural stem cells. iScience. 15, 524-535 (2019).
- Laustsen, A., et al. Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function. Nature Communications. 9 (1), 1-14 (2018).
- Bak, R. O., et al. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. eLife. 6, 27873 (2017).
- Nakauchi, Y., et al. The cell type-specific 5hmC landscape and dynamics of healthy human hematopoiesis and TET2-mutant preleukemia. Blood Cancer Discovery. 3 (4), 346-367 (2022).
- Vaidyanathan, S., et al. selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell. 26 (2), 161-171 (2020).
- Tang, N., et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 5 (4), 133977 (2020).
- Georgiadis, C., et al. Long terminal repeat CRISPR-CAR-coupled "universal" T cells mediate potent anti-leukemic effects. Molecular Therapy. 26 (5), 1215-1227 (2018).
- Ren, J., et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clinical Cancer Research. 23 (9), 2255-2266 (2017).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Doench, J. G., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Bak, R. O., Porteus, M. H. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Reports. 20 (3), 750-756 (2017).
