Enrichment of Adult Mouse Dorsal Root Ganglia Neuron Cultures by Immunopanning
Summary
This paper describes an immunopanning protocol for adult mouse dorsal root ganglia. By adhering antibodies to culture plates, we can negatively select and remove non-neuronal cells. We show that the cultures are enriched for neurons using this protocol, allowing for an in-depth study of neuronal responses to manipulation.
Abstract
Dorsal root ganglia (DRGs) are peripheral structures adjacent to the dorsal horn of the spinal cord, which house the cell bodies of sensory neurons as well as various other cell types. Published culture protocols often refer to whole dissociated DRG cultures as being neuronal, despite the presence of fibroblasts, Schwann cells, macrophages, and lymphocytes. While these whole DRG cultures are sufficient for imaging applications where neurons can be discerned based on morphology or staining, protein or RNA homogenates collected from these cultures are not primarily neuronal in origin. Here, we describe an immunopanning sequence for cultured mouse DRGs. The goal of this method is to enrich DRG cultures for neurons by removing other cell types. Immunopanning refers to a method of removing cell types by adhering antibodies to cell culture dishes. Using these dishes, we can negatively select against and reduce the number of fibroblasts, immune cells, and Schwann cells in culture. This method allows us to increase the percentage of neurons in cultures.
Introduction
Dorsal root ganglia (DRGs) house the cell bodies of the sensory neurons which innervate peripheral tissues. Studying these neurons allows us to understand the mechanistic underpinnings of pain and sensory conditions. However, DRGs are not comprised of neurons alone, but also contain fibroblasts, Schwann cells, macrophages, and other immune cells1. Despite the presence of these various cell types, whole DRG cultures are often referred to in the literature as neuronal2,3. These cultures are still useful for neuronal investigation by imaging or flow cytometry, which would allow for neuronal identification by staining, cell size, and/or morphology. However, for assays such as polymerase chain reaction (PCR) or western blotting, where cultures are homogenized for RNA or protein collection, the presence of non-neuronal cell types may interfere with results. Hence, there is a need to increase the proportion of neuronal cells in DRG cultures.
Immunopanning is a technique used to purify a variety of cell types, including cortical neurons, astrocytes, oligodendrocyte precursor cells, and microglia. In simple words, it involves adhering an antibody against a cell surface marker to a Petri dish, intended to bind certain cells (Figure 1), and it can be used to select either for or against cell types of interest4,5,6. Immunopanning rat embryonic DRGs to select against non-neuronal cells has been described previously by Zuchero7. However, we have been unable to find an immunopanning protocol specific to mouse DRGs. In this protocol, we build on the basic tenants of the Zuchero protocol, but instead adapted the immunopanning sequence to enrich adult mouse DRG cultures (Figure 2). This is a powerful tool to study established sensory neurons in culture. It is advantageous as it allows for the use of adult mice from genetic, disease, and injury models, so that their sensory neurons may be studied with greater specificity.
This protocol is advantageous for readers looking to increase the proportion of neurons in adult mouse DRG cultures. However, the immunopanning steps can also be omitted, and this protocol can also be used for whole DRG cultures.
Protocol
All animal experiments were performed with approval from the University of Alberta Health Sciences Animal Care and Use Committee (protocol 0000274).
1. Reagent preparation
- For day 1, prepare the following reagents: cell culture grade water; poly-D-lysine-prepare a stock by reconstituting the whole bottle in 50 mL of culture grade water to a stock concentration of 100 µg/mL, store at 4 °C, and dilute the stock 1:10 in culture grade water to a final concentration of 10 µg/mL; tris-HCl-prepare a stock by dissolving powdered tris hydrochloride in culture grade water to a stock concentration of 500 mM (3.94 g in 50 mL), pH 9.5, filter sterilize (0.22 µm), store at 4 °C, and dilute the stock 1:10 in culture grade water to a final concentration of 50 mM; secondary antibodies (goat anti-rat, -rabbit, and -mouse).
- For day 2, prepare the following reagents.
- Prepare cell culture grade Ca2+, Mg2+-free Hank's balanced salt solution (HBSS; HBSS-/-), and cell culture grade D-phosphate buffered saline (PBS) with Ca2+ and Mg2+. Prepare a laminin stock aliquot and store at -80 °C immediately upon receiving; dilute the stock in sterile HBSS-/- to a final concentration of 10 µg/mL for working stock.
- Prepare 4% bovine serum albumin (BSA) stock by dissolving 400 mg of BSA in 7.5 mL of D-PBS (pH 7.4), bring the volume to 10 mL, filter sterilize (0.22 µm), aliquot, and store at -20 °C. Prepare 0.2% BSA by diluting 750 µL of 4% BSA in 14.25 mL of D-PBS.
- Prepare 0.4% DNAse stock by adding 1 mL of Earle's balanced salt solution (EBSS) per 12,500 units of DNAse, filter sterilize (0.22 µm), aliquot, and store at -20 °C. Prepare panning buffer (0.02% BSA) by adding 3 mL of 0.2% BSA and 100 µL of 0.4% DNAse in 27 mL of D-PBS. Prepare primary antibodies (CD45, PDGRFβ, O4).
NOTE: This protocol uses an O4 hybridoma8,9, however, 10 µg of a commercially available O4 antibody would also be appropriate. - Prepare the combination collagenase stock by dissolving a bottle of combination collagenase (50 mg) in 5 mL of HBSS-/-, aliquot, and store at -20 °C. Prepare a working stock per two mice by adding 500 µL of warmed combination collagenase and 50 µL of warmed 0.4% DNase to 4.5 mL of HBSS-/-.
- Prepare low ovomucoid by adding 3 g of BSA to 150 mL of D-PBS, then add 3 g of trypsin inhibitor and mix to dissolve. Adjust the pH to 7.4 and bring the volume up to 200 mL with D-PBS. Filter sterilize, prepare 1 mL aliquots, and store at -20 °C. On the day of dissection, combine 1 mL of low ovomucoid in 9 mL of D-PBS for the working solution.
- Prepare 15% BSA by dissolving 7.5 g of BSA in 50 mL of HBSS-/- (place on a shaker to dissolve fully), filter sterilize, and store at 4 °C.
- Prepare 50 mL of neuron media by adding 23.2 mL of DMEM, 23.2 mL of neurobasal medium, 500 µL of glutamax, 500 µL of sodium pyruvate, 500 µL of penicillin/streptomycin (aliquot immediately upon arrival and store at -20 °C) , 500 µL of insulin (5 µg/mL final), 500 µL of SATO, 50 µL of N-acetyl-L-cysteine (NAC; 5 µg/mL final), and 1,000 µL of B27+ (immediately aliquot and store at -20 °C upon receiving).
- Prepare the insulin stock by dissolving in culture grade water to a concentration of 0.5 mg/mL (50 mg in 100 mL), adjust the pH to 7.4, filter sterilize (0.22 µm), prepare 500 µL aliquots, and store at -20 °C.
- Prepare the SATO stock by combining 20 µL of progesterone (2.5 mg in 100 µL of ethanol), 800 µL of sodium selenite (4 mg in 10 mL of neurobasal media + 10 µL of 1 N NaOH), 800 mg of BSA, 800 mg of transferrin, 128 mg of putrescine dihydrochloride, and 80 mL of neurobasal. Filter sterilize (0.22 µm), prepare 500 µL aliquots, and store at -20 °C. It is essential to make fresh progesterone and sodium selenite solutions.
- Prepare 50 µL of N-acetyl-L-cysteine (NAC) stock by dissolving in neurobasal medium to a stock concentration of 5 mg/mL (50 mg in 10 mL), filter sterilize (0.22 µm), aliquot, and store at -20 °C.
- For day 4 and 5, prepare the following reagents.
- Prepare 0.2 M phosphate buffer (PB) by dissolving 21.8 g of sodium phosphate dibasic (Na2HPO4) and 8.34 g of sodium phosphate monobasic dihydrate (NaH2PO4) in 1,000 mL of water. Adjust the pH to 7.4 and store at room temperature.
- Prepare 8% paraformaldehyde (PFA) as follows: in a fume hood, warm 50 mL of water to 55 °C, turn off the heat, and add 8 g of PFA prills, stirring constantly. Add 1 N NaOH dropwise until the solution begins to clear, then add 40 mL of 0.2 M PB, and continue stirring until clear. Cool the solution to room temperature, adjust the pH to 7.4, and bring the volume up to 100 mL with 0.2 M PB. Filter and store at 4 °C for up to 2 weeks.
- Prepare D-PBS + Triton X-100 (PBSTX) by adding 0.2% Triton X-100 in culture grade D-PBS (1 mL of Triton + 500 mL of D-PBS). Store at 4 °C. Prepare blocking solution, which is 1% normal donkey serum (NDS) in PBSTX. Ahead of time, 1 mL of NDS + 9 mL of PBSTX can be prepared and stored at -20 °C in 10 mL aliquots.
- Prepare antibody solution which is 0.2% NDS/0.2% BSA in PBSTX : 200 µL of NDS + 200 mg BSA + 9.6 mL of PBSTX; can be prepared ahead of time and stored at -20 °C in 10 mL aliquots.
2. Equipment setup
- Set up the following equipment: sterile cell culture biosafety cabinet, micropipette set, serological pipettes and pipette gun, 10 cm Petri dishes, desired cell culture dishes for plating (here, 24-well black glass-bottom plates were used for imaging), 15 mL and 50 mL conical tubes, dissection tools (namely: fine spring scissors for laminectomy and nerve trimming, fine forceps, and a razor blade), water bath set to 37 °C, 70 µm cell strainer, centrifuge (10 min at 300 x g) with adaptors for 15 mL conical tubes, trypan blue and haemocytometer, fume hood, and magnetic stirring hot plate.
3. Experimental method
- Day 1: preparation of dishes
- In a biosafety cabinet, coat the desired culture surface with enough poly-D-lysine to cover the bottom of the well. Store overnight at 4 °C. Here, 24-well glass-bottom plates were used for imaging, and thus 300 µL of poly-D-lysine was used.
- In a biosafety cabinet, prepare the panning dishes: for the CD45 dish, add 10 mL of working tris-HCl solution and 30 µL of goat anti-rat IgG to a 10 cm Petri dish; for the PDGFRβ dish, add 10 mL of working tris-HCl solution and 30 µL of goat anti-rabbit IgG to a 10 cm Petri dish; for the O4 hybridoma dish, add 10 mL of working tris-HCl solution and 30 µL of goat anti-mouse IgG to a 10 cm Petri dish. Ensure the solution covers the entire dish area and leave at 4 °C overnight.
- Day 2: dissection, trituration, and immunopanning
- Aspirate the poly-D-lysine, wash the plates three times with tissue culture water, tilt the lid open, and allow the surface to dry completely in a biosafety cabinet. Apply enough laminin to the plates to coat the bottom of each well, and place in an incubator at 37 °C. For the 24-well plates, 200 µL is sufficient.
- Finish preparing the panning dishes. Wash each dish with D-PBS and incubate with 10 µg of primary antibody. Allow to sit at room temperature for at least 2 h.
- For the CD45 dish, add 5 mL of 0.2% BSA and 20 µL of CD45 primary; for the PDGFRβ dish, add 5 mL of 0.2% BSA and 15.4 µL of PDGFRβ; for the O4 dish, add 3 mL of 0.2% BSA, 2 mL of O4 hybridoma, and an additional 100 µL of 4% BSA (to ensure the final BSA concentration remains at 0.2%).
- Euthanize one to four mice using 0.1 mL/kg sodium pentobarbitol (per 10 cm immunopanning dish). We used C57BL/6 mice at 8-10 weeks old. Both sexes were used and cultured separately from one another. Perfuse the animal with 30 mL of cold 0.9% saline.
- Pin the front and hind paws to a styrofoam stage and use a clean razor blade to expose the spinal column from the back. Perform a laminectomy by making cuts about half-way deep on either side of the dorsal spinal column, removing the dorsal half of the column, to expose the spinal cord. Either gently cut the nerves and remove the spinal cord, or gently push the cord to the side, maintaining the nerve integrity.
- Use the spinal nerve roots (either severed or attached to the spinal cord) to find and remove all the DRGs from either side of the spinal column. Trim the nerve debris from each DRG as it is removed (more nerve debris in the culture may lead to poor culture survival). Collect the DRGs in 15 mL of HBSS-/- in a 15 mL conical tube on ice.
NOTE: Note that the tissue is dissected in open air, but once the DRGs are added to the tube they should be treated as sterile and only manipulated in a biosafety cabinet. - Place frozen stocks of stemxyme 1 and 0.4% DNAse in a water bath, approximately 30 min prior to the end of the dissections (roughly when starting the last mouse). Allow them to settle to the bottom of the conical tube, then continue the protocol in a biosafety cabinet.
- Aspirate most of the HBSS-/- from the DRGs. Use a pipette rather than suction to get <1 mL of liquid, and leave ~100 µL liquid so as not to risk losing tissue.
- Wash three times with 1 mL of HBSS-/-. Add 5 mL of working stemxyme solution to the tissue. Cover the lid with a transparent film and float the tube on its side in a water bath set to 37 °C for 1 h.
- After incubation in the enzyme, add 1 mL of low ovo inhibitor solution to the tube. Triturate the cells gently with a p1000 pipette 10-15 times. Allow the tissue chunks to settle.
- Transfer the top 2-3 mL of solution (containing dissociated cells) to fresh low ovomucoid solution. Repeat until the tissue is fully dissociated and no visible chunks remain.
- Centrifuge for 10 min at 300 x g at room temperature. Siphon off the supernatant again, using a pipette rather than suction for the last 1 mL, leaving ~100 µL, and resuspend the cell pellet in 1 mL of panning buffer.
- Gently pipette to mix. Pre-wet a 70 µm cell strainer with 1 mL of panning buffer over a 50 mL conical tube. Filter the cell solution through the cell strainer.
- Wash the tube with 1 mL of panning buffer, then pass through the strainer (3 mL of filtrate). Layer the cell suspension gently (1 mL at a time) on top of 2 mL of a 15% BSA cushion to remove myelin debris. A 2 mL cushion is effective for two mice, and 3 mL is effective for four mice.
- For layering, place the 2 mL of BSA in the tube, close, and gently coat the sides of the tube with BSA. Then, gently layer the cell suspension of top, by pipetting against the side of the tube.
- Centrifuge at room temperature at 300 x g for 10 min (slow acceleration and deceleration). Use a P1000 pipette to remove the clear liquid on top, the myelin phase in between, and the BSA, 1 mL at a time (changing tips in between each mL). Leave ~100 µL of BSA so as not to disturb the pellet.
- Add 5 mL of panning buffer and incubate the tube in a 37 °C, 10% CO2 incubator for 30-45 min. This allows for antigen retrieval. Be sure to loosen the cap to allow gas exchange.
- Wash the CD45 dish three times with D-PBS. Pour off the final rinse. Decant the cell suspension into the CD45 dish.
- Incubate the cells on the CD45 dish for a total of 20 min at room temperature, swirling gently at the 10 min point to allow the cells equal access to the antibody.
- Rinse the PDGFRβ dish three times with D-PBS and pour off the final rinse. Transfer the unbound cells from the CD45 dish to the PDGFRβ dish.
- Gently shake the CD45 dish and prop it up at an angle. Gently pipette 1 mL of the cell suspension over the dish to collect unbound cells. Decant it into the PDGFRβ dish and pipette to transfer the remaining cell suspension to the PDGFRβ plate.
- Incubate the PDGFRβ plate for a total of 20 min at room temperature, swirling gently at the 10 min point to allow the cells equal access to the antibody.
- Rinse the O4 dish three times with D-PBS and pour off the final rinse. Transfer the unbound cells from the PDGFRβ dish to the O4 dish using the same method as in step 3.2.19. Incubate the O4 plate for a total of 20 min at room temperature, swirling gently at the 10 min point to allow the cells equal access to the antibody.
- Transfer the cell suspension to a 15 mL conical tube and centrifuge for 10 min at 300 x g. Resuspend the cells in the desired volume, and dilute 1:1 with trypan blue for cell viability. Count medium to large cells with a haemocytometer (this will allow for the best representation of the number of neurons in the culture).
- Remove the laminin and plate the cells at the desired density. Do not allow plate to dry between laminin removal and plating, and add the cells immediately.
- Culture the quantified cells for neuronal enrichment (Figure 3) with 500 neurons in 25 µL of culture medium (described in 1.2.7) per well in 24-well plates and incubate at 37 °C for 30 min. Flood the wells to a final volume of 500 µL.
- Day 4: immunocytochemistry (ICC) fixation, blocking, and primary
- After 48 h of growth, add 500 µL/well (or volume equivalent to the media in the well) of 8% PFA for 15 min at room temperature to fix the cells. If media analysis is desired, one may fix with 4% PFA directly. However, we have not tested this.
- Wash the cells with D-PBS three times. Note, we have kept these cells in culture without media change for a maximum of 72 h, but hypothesize they could survive longer, particularly if the media is half-changed out.
- Remove the final wash and add enough blocking solution per well to cover the cells completely. Block for 30 min at room temperature. Remove the blocking solution and add primary antibody in antibody solution: β3-tubulin at a 1:1,000 dilution. Seal the plate with transparent film and incubate overnight at 4 °C.
NOTE: One might consider staining with PDGFRβ for the absence of fibroblasts, CD45 for the absence of immune cells, P0 or PMP22 for the absence of myelin, or fabp7 for satellite glia.
- Day 5: ICC secondary
- Wash the cells with D-PBS three times. Remove the final wash and add secondary antibodies in antibody solution: anti-rabbit 488 at a 1:500 dilution and DAPI at a 1:2,000 dilution. Incubate for 30 min at room temperature, protected from light.
- Wash with D-PBS three times. Add enough D-PBS to cover the bottom of the well, and image. The plate can be sealed with transparent film, protected from light, and stored at 4 °C for up to 2 weeks.
Representative Results
The fixed cells stained with DAPI and β3-tubulin were then imaged with a confocal high content screening system. Images were analyzed using suitable commercial software to determine the percentage of DAPI-positive cells that co-labelled with β3-tubulin (Figure 3). Whole DRG cultures were determined to have 42.36% ± 6.4% β3-tubulin staining, and immunopanned DRG cultures were determined to have 71.44% ±7.43% β3-tubulin staining. This reveals a significant increase in neuronal enrichment with immunopanning (p < 0.0001, unpaired t-test).
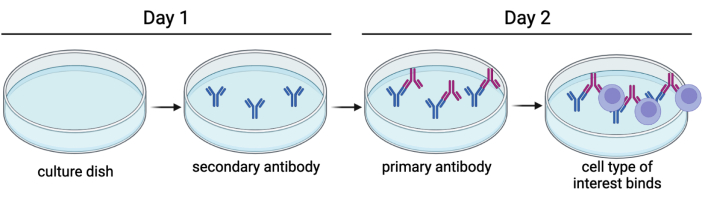
Figure 1: Immunopanning basics. A secondary antibody is adhered to a culture dish overnight, and primary antibodies are then introduced. This allows the cells of interest to be attached to the plate by a surface antigen. Please click here to view a larger version of this figure.
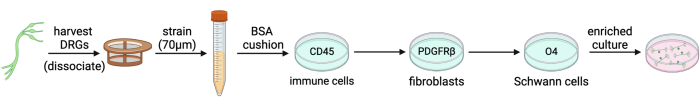
Figure 2: Adult mouse DRG neuron immunopanning by negative selection. DRGs are harvested, dissociated, and strained into a single cell solution. Myelin debris is removed by centrifugation through a BSA cushion. The cell suspension is then passed across three immunopanning dishes (CD45, PDGFRβ, and O4) to achieve a culture that is enriched for neurons. Please click here to view a larger version of this figure.
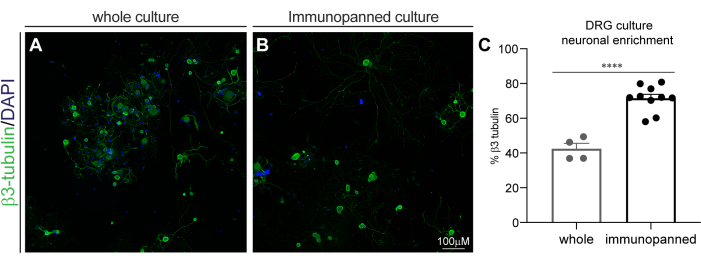
Figure 3: DRG culture neuronal enrichment by immunopanning. Fixed cultures were stained with β3-tubulin for neurons, and DAPI for all nuclei. (A) Representative image of a whole (non-immunopanned) culture. (B) Representative image of an immunopanned culture. (C) Quantification of β3-tubulin-positive cells as a percentage of total DAPI-positive cells. Bars indicate mean ± standard error mean (SEM). **** = p < 0.0001, unpaired t-test. Please click here to view a larger version of this figure.
Discussion
This immunopanning protocol increases the proportion of neuronal cells in DRG primary cultures. For the best results, dissections should be done in a timely manner, and DRGs should be trimmed of excess nerves. The dissociation step should be carefully monitored, and should not exceed 1 h, to prevent the cells from unnecessary stress. With regard to the immunopanning specifically, each plate should be gently swirled at the halfway point to allow the cells to have access to the coated antibodies. The plates should also be viewed under a culture microscope after the neuronal cell suspension is collected to ensure that non-neuronal cells have bound.
A limitation in the use of adult mice is the established nature of the DRGs. An inability to remove all non-neuronal cell types leaves roughly 30% of the culture as non-neuronal (Figure 3). While we can dissociate and reduce the population of immune cells, fibroblasts, and Schwann cells from the cultures, satellite glia may be exceedingly difficult to dissociate from neurons. We also hypothesize that these neurons could have poor survival without the metabolic support provided by satellite glia10. While our attempt to stain for these cells using GFAP failed due to poor antibody specificity, a future direction may be to stain for these cells using fabp7. We attempted to remove CD9-positive non-neuronal cells such as satellite glia when troubleshooting this protocol. However, in the CD9 plate, it was found that many neurons were attached to the plate. We hypothesize that mouse neurons may have some CD9 expression. In fact, extracellular vesicle literature from mouse neuron-like cells employs CD9 as a marker11. If a more suitable panning antibody could be found to reduce the proportion of satellite glia, one might consider additional supplementation with growth factors such as NGF, BDNF, or NT3 to counteract the loss of these supporting cells.
This immunopanning protocol can improve the accuracy of neuronal readouts from homogenous DRG culture samples by increasing the proportion of neuronal cells in culture. It also provides an avenue to culture adult mouse DRG neurons, allowing for an in-depth investigation of neuronal phenotypes in genetic, disease, and injury mouse models.
Immunopanning is also a highly customizable tool. For example, the cell surface marker isolectin-B4 might be employed to select for non-peptidergic mouse nociceptors specifically. Immunopanning can also be used to enrich other primary cultures, including cortical neurons, microglia, astrocytes, and oligodendrocyte precursor cells. It is a powerful tool that can broaden the applications of primary cell cultures.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by a Project Grant from the Canadian Institutes of Health Research (FRN-162434) and a Discovery Grant from the MS Society of Canada (EGID-3761). The authors would like to thank Dr. Sun and the Cross Cancer Institute for training and use of the ImageXpress system.
Materials
| 0.2 um Syringe filters | Fisher | 723-2520 | |
| 100 mm petri dishes | ThermoFisher | FB0875712 | |
| 24 well black glass-bottom plates | Cellvis | P24-1.5H-N | |
| 70 um cell strainer | Cedarlane | 15-1070-1(BI) | |
| B27+ supplement | Gibco | A3582801 | |
| Bovine Serum Albumin | Sigma Aldrich | A7906 | for 15% BSA cushion, and ICC (heat shock fraction ≥98%) |
| Bovine Serum Albumin | Sigma Aldrich | A4161 | for 4% BSA for immunopanning, and SATO for media (essentially globulin free, suitable for cell culture, ≥99%) |
| CD45 antibody | BD Pharmigen | 550539 | |
| DAPI | Invitrogen | D1306 | |
| DMEM | Gibco | 11960069 | |
| DNAse | Worthington | LS002007 | for stemxyme solution and panning buffer |
| D-PBS | Sigma Aldrich | D8662 | |
| EBSS | Sigma Aldrich | E6267 | for DNAse solution |
| Filter paper P8 grade | ThermoFisher | 09-795K | for 8% PFA |
| Glutamax | ThermoFisher | 35050061 | |
| goat-anti-mouse IgG | Jackson Immunoresearch | 115-005-020 | for O4 dish |
| goat-anti-rabbit 488 | Invitrogen | A11008 | |
| goat-anti-rabbit IgG | Jackson Immunoresearch | 115-005-003 | for PDGFRB dish |
| goat-anti-rat IgG | Jackson Immunoresearch | 115-005-044 | for CD45 dish |
| HBSS -/- | Sigma Aldrich | 14175145 | |
| Insulin | Sigma Aldrich | I2643 | |
| laminin | Invitrogen | 23017-015 | |
| N-acetyl cysteine | Sigma Aldrich | A8199 | |
| Neurobasal | Gibco | 21103049 | |
| Normal Donkey Serum | Sigma-Aldrich | D9663 | |
| O4 antibody | n/a | n/a | Hybridoma |
| Ovomucoid trypsin inhibitor | Cedarlane | LS003086 | for low ovo |
| paraformaldehyde prills | Sigma Aldrich | 441244 | for 8% PFA |
| PDGFRB antibody | Abcam | AB32570 | |
| penicillin/ streptomycin | Gibco | 15140-122 | |
| Poly-D-Lysine | Sigma Aldrich | P6407 | |
| Progesterone | Sigma Aldrich | P8783 | for SATO |
| Putrescine dihydrochrloride | Sigma Aldrich | P5780 | for SATO |
| Sodium phosphate dibasic | Fisher | S374-1 | for 0.2 M PB |
| Sodium Phosphate monobasic dihydrate | Sigma Aldrich | 04269 | for 0.2 M PB |
| Sodium Pyruvate | ThermoFisher | 11360070 | |
| Sodium Selenite | Sigma Aldrich | S5261 | for SATO |
| Stemxyme I | Cedarlane | LS004107 | for tissue dissociation; combination collagenase |
| Transferrin | Sigma Aldrich | T1147 | for SATO |
| Tris-HCl | Millipore Sigma | T5941 | |
| trypan blue | Gibco | 15250-061 | |
| β3-Tubulin | Sigma-Aldrich | T2200 |
References
- Haberberger, R. V., Barry, C., Dominguez, N., Matusica, D. Human dorsal root ganglia. Frontiers in Cellular Neuroscience. 13, 271 (2019).
- Perner, C., Sokol, C. L. Protocol for dissection and culture of murine dorsal root ganglia neurons to study neuropeptide release. STAR Protocols. 2 (1), 100333 (2021).
- Lin, Y. T., Chen, J. C. Dorsal root ganglia isolation and primary culture to study neurotransmitter release. Journal of Visualized Experiments:JoVE. (140), e57569 (2018).
- Foo, L. C., et al. Development of a method for the purification and culture of rodent astrocytes. Neuron. 71 (5), 799-811 (2011).
- Nolle, A., et al. Enrichment of Glial Cells From Human Post-mortem Tissue for Transcriptome and Proteome Analysis Using Immunopanning. Frontiers in Cellular Neuroscience. 15, 772011 (2021).
- Barres, B. A. Designing and troubleshooting immunopanning protocols for purifying neural cells. Cold Spring Harbor Protocols. 2014 (12), 1342-1347 (2014).
- Zuchero, J. B. Purification of dorsal root ganglion neurons from rat by immunopanning. Cold Spring Harbor Protocols. 2014 (8), 826-838 (2014).
- Sommer, I., Schachner, M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. 발생학. 83 (2), 311-327 (1981).
- Sommer, I., Schachner, M. Cell that are O4 antigen-positive and O1 antigen-negative differentiate into O1 antigen-positive oligodendrocytes. Neuroscience Letters. 29 (2), 183-188 (1982).
- Hanani, M. Satellite glial cells in sensory ganglia: from form to function. Brain Research. Brain Research Reviews. 48 (3), 457-476 (2005).
- Viveiros, A., et al. In-cell labeling coupled to direct analysis of extracellular vesicles in the conditioned medium to study extracellular vesicles secretion with minimum sample processing and particle loss. Cells. 11 (3), 351 (2022).
