To verify the validity of the proposed protocol, the PD experiments presented here were performed with a biotinylated RNA aptamer designed in silico to specifically bind TDP-4320. This RNA binds its protein target with high binding affinity (Kd = 90 nM)20. Here, this RNA, of sequence 5'-CGGUGUUGCU-3', is referred to with the name "+RNA". As a negative control, the reverse complementary sequence of +RNA, which is here called "-RNA", was used. Its sequence is 5'-AGCAACACCG-3'. -RNA shows a significantly lower binding affinity toward TDP-43 (Kd = 1.5 µM)19. For the purpose of the protocol described here, these RNA oligonucleotides have been purchased conjugated to a biotin molecule, to allow binding to the streptavidin beads. +RNA was purchased with a biotin-TEG at its 3' end, which includes a 15-atom triethylene glycol spacer between the biotin and the phosphate group of the nucleic acid; -RNA instead had a biotin at its 5' end, conjugated to the nucleic acid via an amino-C6 linker. However, if the design of the RNA bait is robust, and as long as there is no structural or chemical interference between the linker and the RNA, other positions for the biotin conjugation and other linker lengths could be employed.
Knowing the identity of the main protein to be found bound to the +RNA probe after the PD enabled the validation of the protocol by identification of TDP-43 in the eluate, using both mass spectrometry (MS) and western blot (WB) (Figure 1).
MS analysis was carried out on four PD replicates performed using either +RNA or -RNA (Figure 2). The identification of the interactomes of +RNA and -RNA is beyond the scope of this protocol, however some results that validate the accuracy of the protocol are reported. Of note, plotting the significantly enriched proteins in a volcano plot revealed that the total protein content and the enriched proteins eluted from +RNA was significantly higher that what was recovered from -RNA (Figure 2). This means that, despite having the same length and structural content (linear), +RNA can establish a higher number of specific interactions, which are retained up to the elution step with high salt. It is likely that -RNA instead establishes a higher number of nonspecific contacts that are disrupted during the washing steps. As expected, TDP-43 was identified as a unique interactor of +RNA20; the average label-free quantification (LFQ) for the four PD replicates performed with +RNA is 31.96 ± 0.56, while the protein is not identified among the interactors of -RNA. In addition, among all unique interactors of +RNA, TDP-43 was found to be the most abundantly enriched protein.
To further validate the protocol, the in-house algorithm catRAPID18,19 was used to computationally predict which other proteins would specifically bind either +RNA or -RNA. In particular, interaction scores for +RNA and -RNA with the proteins composing the human proteome were computed using the catRAPID 'interaction propensity' feature, as defined in our previous work27. Among the proteins scored with high confidence, HNRNPH3 was predicted to bind selectively +RNA (+RNA interaction score = 1.01; -RNA interaction score = 0.21) and PCBP2 to interact specifically with -RNA (+RNA interaction score = -0.5; -RNA interaction score = 0.31) (Figure 3A). In addition, the protein RBM41 was predicted to be promiscuous for both RNA oligonucleotides (+RNA interaction score = 0.4; -RNA interaction score = 0.39) (Figure 3A). The MS analysis indeed confirmed the presence of HNRNPH3 and PCBP2 in the PD of +RNA and -RNA respectively, while RBM41 was found interacting with both (Figure 3B).
WB was used to detect the presence of TDP-43 to further confirm the results and during protocol optimization (Figure 4). In the procedure described here, different samples were collected at different stages. The input sample (IN) consisted of the total proteins diluted in lysis buffer. The flowthrough (FT) was obtained after an overnight incubation of the total proteins with the streptavidin beads pre-coated with the biotinylated RNA, representing the fraction of proteins that did not bind the RNA. Finally, the eluate (EL) contained all the proteins that recognized specifically the RNA under investigation, since between the FT and the EL steps three washing steps with 150 mM salt and 0.1% triton-X should have removed the weakest interactions.
For each replicate, the same amount (5% v/v) of IN, FT, and EL was run in parallel on an SDS-PAGE and stained with an anti-TDP-43 antibody (Figure 4). In the case of +RNA, the band of TDP-43 was observed in IN and in EL, indicating that the protein, present from the start in the total protein extract, is retained by +RNA during the washing steps and is only eluted at the end with a high salt buffer. TDP-43 was also present in IN for -RNA, however the band corresponding to the protein is also visible in FT, indicating that this RNA does not bind TDP-43. The absence of the TDP-43 band in EL confirms this result.
During the optimization of the protocol, the elution of the proteins specifically bound to the RNA sequences was probed both with an elution buffer containing 1 M NaCl (EB1) and with an elution buffer complete with 2 M NaCl (EB2) (Figure 4). Eluates obtained with either EB were compared on an SDS-PAGE and blotted with the anti-TDP-43 antibody. The images obtained were then analyzed with ImageJ28 to quantify any difference in TDP-43 amount eluted with the two buffers. Overall, no significant difference was observed, and we concluded that, within these assays, 1 M salt is sufficient to disrupt even the strongest protein-RNA interactions.
Overall, the results reported here for MS and WBs demonstrate that this protocol is efficient in capturing the protein interactors of a given RNA in a specific manner, and that it enables the elution in buffers compatible with downstream analysis.
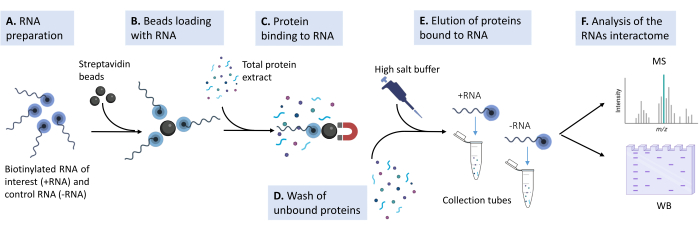
Figure 1: Sketch of the experimental pipeline used in the proposed protocol. (A) The biotinylated RNA oligonucleotide is prepared in lysis buffer at the appropriate concentration. (B) Magnetic streptavidin beads are washed, blocked with yeast tRNA, and loaded with the biotinylated RNA. (C) Total protein extract derived from cultured mammalian cell lines are added to the beads-RNA mixture. (D) Multiple washes are performed to remove nonspecific interactions. (E) The specific protein interactors are detached from the RNA with a hypertonic solution. (F) The identity of the interactors is revealed by mass spectrometry, and specific cases are validated by western blot. Please click here to view a larger version of this figure.
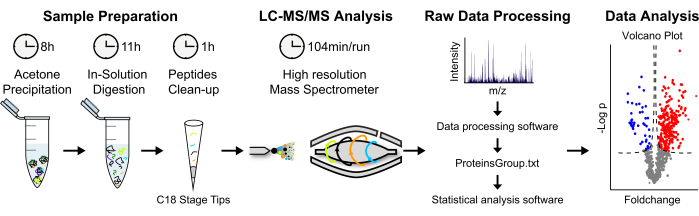
Figure 2: Analytical strategy for label-free MS-based protein quantification. (A) Eluted proteins are precipitated in cold acetone overnight. Proteins are then denatured, and an in-solution digestion is performed. Proteolytic peptides are concentrated and desalted. (B) Peptides are analyzed via LC-MS/MS using a "shotgun approach". (C) Raw data processing and analysis is performed using MaxQuant and Perseus software, respectively. (D) Statistically significant enriched proteins are displayed in a volcano plot. Please click here to view a larger version of this figure.
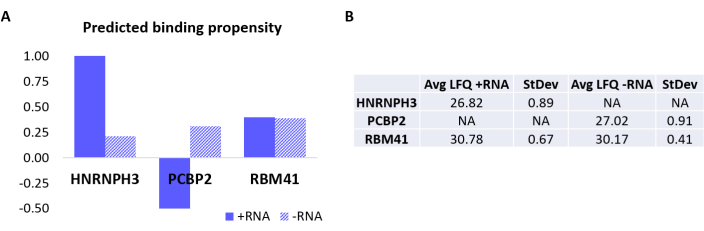
Figure 3: Correlation between predicted interaction propensities and experimentally determined interactions of +RNA and -RNA. (A) catRAPID interaction scores relative to HNRNPH3, PCBP2, and RBM41, indicating preferential binding of HNRNPH3 for +RNA and of PCBP2 for -RNA, while RBM41 is predicted to indiscriminately bind both RNA sequences. (B) Label-free quantification averages determined by mass-spectrometry analysis from the pull-downs performed with +RNA and -RNA. The analysis confirms that HNRNPH3 solely binds +RNA, PCBP2 solely binds -RNA, and RBM41 binds both equally. Please click here to view a larger version of this figure.
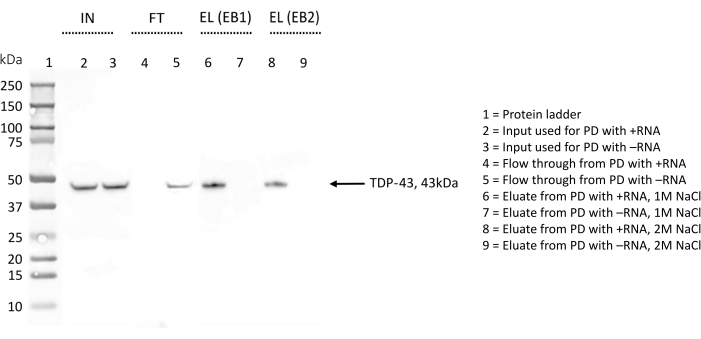
Figure 4: Western blot validation of the presence/absence of TDP-43 among the interactors of chosen RNA sequences. The WB membrane has been treated with anti-TDP-43 antibody. IN = input; FT = flow-through; EL (EB1) = elution with elution buffer 1; EL (EB2) = elution with elution buffer 2; the sign "+" indicates samples derived from the pull-down performed with +RNA; the sign "-" indicates samples derived from the pull-down performed with -RNA; lane 1 contains a protein ladder. TDP-43 is indicated by an arrow. The WB indicates that TDP-43 is found among +RNA interactors but not among -RNA interactors. Please click here to view a larger version of this figure.
| Buffer Name | Composition | |||||
| 10x Tranfer buffer | 250 mM tris, 1.92 M glycine, 1% SDS, 20% methanol. Dilute 10 folds prior use | PD | ||||
| 20X MES SDS running buffer | 1 M MES, 1 M tris, 2% SDS , 20 mM EDTA. Adjust pH to 7.3. Dilute 20 folds prior use | |||||
| 4x Sample loading buffer | 0.25 M Tris base, 0.28 M SDS, 40% glycerol, 20% 2-mercapto-ethanol, 4 mg/ml bromphenol blue | |||||
| Elution buffer 1 | 20 mM phosphate pH 7.5, 1 M NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT (to be added after quantification) | |||||
| Elution buffer 2 | 20 mM phosphate pH 7.5, 2 M NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT (to be added after quantification) | |||||
| Lysis buffer | 10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT and protease inhibitors | |||||
| Tris-buffered saline with Tween-20 | 1 M Tris-HCl pH 7.4, 3 M NaCl, 2.0% Tween-20 | |||||
| Wash buffer 1 | 10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5 mM EDTA, 0.1 % Triton TM X-100, 1 mM DTT and protease inhibitors | |||||
| Wash buffer 2 | 25 mM Hepes pH 8, 150 mM NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT and protease inhibitors | |||||
| Buffer A | 0.1% formic acid | MS | ||||
| Buffer B | 60% acetonitrile, 0.1% formic acid | |||||
| Denaturation buffer | 8M urea, 50 mM Tris-HCl | |||||
Table 1: PD and MS buffers. Names and composition of the buffers used for either the pull-down experiments (PD) or for the mass-spectrometry analysis (MS).
