Transcriptoom bestaat uit de expressies van alle genen in een monster en kan worden geprofileerd door high-throughput technologieën zoals microarray en RNA-seq1. De expressieniveaus van één gen in een dataset worden een transcriptomisch kenmerk genoemd, en de differentiële representatie van een transcriptomisch kenmerk tussen het fenotype en de controlegroep definieert dit gen als een biomarker van dit fenotype 2,3. Transcriptomische biomarkers zijn op grote schaal gebruikt bij het onderzoeken van ziektediagnose4, biologisch mechanisme5 en overlevingsanalyse 6,7, enz.
Genactiviteitspatronen in de gezonde weefsels bevatten cruciale informatie over het leven 8,9. Deze patronen bieden inzichten van onschatbare waarde en fungeren als ideale referenties voor het begrijpen van de complexe ontwikkelingstrajecten van goedaardige aandoeningen10,11 en dodelijke ziekten12. Genen interageren met elkaar en transcriptomen vertegenwoordigen de uiteindelijke expressieniveaus na hun gecompliceerde interacties. Dergelijke patronen worden geformuleerd als transcriptioneel regulatienetwerk13 en metabolismenetwerk14, enz. De expressies van boodschapper-RNA’s (mRNA’s) kunnen transcriptioneel worden gereguleerd door transcriptiefactoren (TF’s) en lange intergene niet-coderende RNA’s (lincRNA’s)15,16,17. Conventionele differentiële expressieanalyse negeerde dergelijke complexe geninteracties met de aanname van onafhankelijkheid tussen kenmerken 18,19.
Recente ontwikkelingen op het gebied van grafische neurale netwerken (GNN’s) tonen een buitengewoon potentieel aan bij het extraheren van belangrijke informatie uit OMIC-gebaseerde gegevens voor kankerstudies20, bijvoorbeeld het identificeren van co-expressiemodules21. De aangeboren capaciteit van GNN’s maakt ze ideaal voor het modelleren van de ingewikkelde relaties en afhankelijkheden tussen genen22,23.
Biomedische studies richten zich vaak op het nauwkeurig voorspellen van een fenotype ten opzichte van de controlegroep. Dergelijke taken worden gewoonlijk geformuleerd als binaire classificaties 24,25,26. Hier worden de twee klasselabels meestal gecodeerd als 1 en 0, waar en onwaar, of zelfs positief en negatief27.
Deze studie had tot doel een gebruiksvriendelijk protocol te bieden voor het genereren van de transcriptionele regulatie (mqTrans)-weergave van een transcriptoomdataset op basis van het vooraf getrainde graph-attention network (GAT)-referentiemodel. Het multitask GAT-framework van een eerder gepubliceerd werk26 werd gebruikt om transcriptomische functies te transformeren naar de mqTrans-functies. Een grote dataset van gezonde transcriptomen van het Xena-platform28 van de University of California, Santa Cruz (UCSC) werd gebruikt om het referentiemodel (HealthModel) vooraf te trainen, dat kwantitatief de transcriptievoorschriften van de regulerende factoren (TF’s en lincRNA’s) tot de doel-mRNA’s heeft gemeten. De gegenereerde mqTrans-weergave kan worden gebruikt om voorspellingsmodellen te bouwen en donkere biomarkers te detecteren. Dit protocol maakt gebruik van de patiëntdataset colonadenocarcinoom (COAD) uit de database van The Cancer Genome Atlas (TCGA)29 als illustratief voorbeeld. In deze context worden patiënten in stadium I of II gecategoriseerd als negatieve monsters, terwijl patiënten in stadium III of IV als positieve monsters worden beschouwd. De verdelingen van donkere en traditionele biomarkers over de 26 TCGA-kankertypes worden ook vergeleken.
Beschrijving van de HealthModel-pijplijn
De methodologie die in dit protocol wordt gebruikt, is gebaseerd op het eerder gepubliceerde raamwerk26, zoals geschetst in figuur 1. Om te beginnen moeten gebruikers de invoergegevensset voorbereiden, deze invoeren in de voorgestelde HealthModel-pijplijn en mqTrans-functies verkrijgen. Gedetailleerde instructies voor het opstellen van gegevens zijn te vinden in hoofdstuk 2 van het protocolgedeelte. Vervolgens hebben gebruikers de mogelijkheid om mqTrans-functies te combineren met de originele transcriptomische functies of alleen door te gaan met de gegenereerde mqTrans-functies. De geproduceerde dataset wordt vervolgens onderworpen aan een functieselectieproces, waarbij gebruikers de flexibiliteit hebben om hun voorkeurswaarde voor k te kiezen in k-voudige kruisvalidatie voor classificatie. De primaire evaluatiemaatstaf die in dit protocol wordt gebruikt, is nauwkeurigheid.
HealthModel26 categoriseert de transcriptomische kenmerken in drie verschillende groepen: TF (transcriptiefactor), lincRNA (lang intergeen niet-coderend RNA) en mRNA (boodschapper-RNA). De TF-kenmerken worden gedefinieerd op basis van de annotaties die beschikbaar zijn in de Human Protein Atlas30,31. Dit werk maakt gebruik van de annotaties van lincRNA’s uit de GTEx-dataset32. Genen die behoren tot de routes op het derde niveau in de KEGG-database33 worden beschouwd als mRNA-kenmerken. Het is vermeldenswaard dat als een mRNA-kenmerk regulerende rollen vertoont voor een doelgen, zoals gedocumenteerd in de TRRUST-database34, het opnieuw wordt geclassificeerd in de TF-klasse.
Dit protocol genereert ook handmatig de twee voorbeeldbestanden voor de gen-ID’s van regulerende factoren (regulatory_geneIDs.csv) en doel-mRNA (target_geneIDs.csv). De paarsgewijze afstandsmatrix tussen de regulerende kenmerken (TF’s en lincRNA’s) wordt berekend door de Pearson-correlatiecoëfficiënten en geclusterd door de populaire tool weighted gene co-expression network analysis (WGCNA)36 (adjacent_matrix.csv). Gebruikers kunnen de HealthModel-pijplijn rechtstreeks gebruiken in combinatie met deze voorbeeldconfiguratiebestanden om de mqTrans-weergave van een transcriptomische gegevensset te genereren.
Technische details van HealthModel
HealthModel geeft de ingewikkelde relaties tussen TF’s en lincRNA’s weer als een grafiek, waarbij de invoerfuncties dienen als de hoekpunten die worden aangeduid met V en een intervertex-randmatrix die wordt aangeduid als E. Elk monster wordt gekenmerkt door K-regulerende kenmerken, gesymboliseerd als VK×1. In het bijzonder omvatte de dataset 425 TF’s en 375 lincRNA’s, wat resulteerde in een monsterdimensionaliteit van K = 425 + 375 = 800. Om de randmatrix E vast te stellen, werd voor dit werk gebruik gemaakt van de populaire tool WGCNA35. Het paarsgewijze gewicht dat twee hoekpunten met elkaar verbindt, weergegeven als 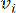 en
en 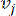 , wordt bepaald door de Pearson-correlatiecoëfficiënt. Het genregulerende netwerk vertoont een schaalvrije topologie36, gekenmerkt door de aanwezigheid van hub-genen met een cruciale functionele rol. We berekenen de correlatie tussen twee objecten of hoekpunten, en , met behulp van de topologische overlapmaat (TOM) als volgt:
, wordt bepaald door de Pearson-correlatiecoëfficiënt. Het genregulerende netwerk vertoont een schaalvrije topologie36, gekenmerkt door de aanwezigheid van hub-genen met een cruciale functionele rol. We berekenen de correlatie tussen twee objecten of hoekpunten, en , met behulp van de topologische overlapmaat (TOM) als volgt:
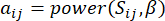 (1)
(1)
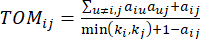 (2)
(2)
De zachte drempel β wordt berekend met behulp van de functie ‘pickSoft Threshold’ uit het WGCNA-pakket. De machtexponentiële functie aij wordt toegepast, waarbij 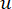 een gen exclusief i en j wordt weergegeven, en
een gen exclusief i en j wordt weergegeven, en 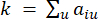 de hoekpuntconnectiviteit wordt weergegeven. WGCNA clustert de expressieprofielen van de transcriptomische kenmerken in meerdere modules met behulp van een veelgebruikte ongelijkheidsmaat (
de hoekpuntconnectiviteit wordt weergegeven. WGCNA clustert de expressieprofielen van de transcriptomische kenmerken in meerdere modules met behulp van een veelgebruikte ongelijkheidsmaat (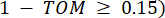 37.
37.
Het HealthModel-raamwerk is oorspronkelijk ontworpen als een multitask-leerarchitectuur26. Dit protocol maakt alleen gebruik van de pre-trainingstaak van het model voor de constructie van de transcriptomische mqTrans-weergave. De gebruiker kan ervoor kiezen om het vooraf getrainde HealthModel verder te verfijnen onder het multitask graph attention network met aanvullende taakspecifieke transcriptomische voorbeelden.
Technische details van de selectie en classificatie van functies
De functieselectiepool implementeert elf algoritmen voor functieselectie (FS). Onder hen zijn er drie op filters gebaseerde FS-algoritmen: het selecteren van de beste kenmerken van K met behulp van de maximale informatiecoëfficiënt (SK_mic), het selecteren van K-kenmerken op basis van de FPR van MIC (SK_fpr) en het selecteren van K-kenmerken met het hoogste percentage valse detectie van MIC (SK_fdr). Daarnaast beoordelen drie op bomen gebaseerde FS-algoritmen individuele kenmerken met behulp van een beslissingsboom met de Gini-index (DT_gini), adaptieve versterkte beslissingsbomen (AdaBoost) en willekeurig bos (RF_fs). De pool bevat ook twee wrapper-methoden: recursieve functie-eliminatie met de lineaire ondersteuningsvectorclassificatie (RFE_SVC) en recursieve functie-eliminatie met de logistische regressieclassificatie (RFE_LR). Ten slotte zijn er twee inbeddingsalgoritmen opgenomen: lineaire SVC-classificatie met de hoogst gerangschikte L1-functiebelangrijkheidswaarden (lSVC_L1) en logistische regressieclassificatie met de hoogst gerangschikte L1-functiebelangrijkheidswaarden (LR_L1).
De classificatiepool maakt gebruik van zeven verschillende classificaties om classificatiemodellen te bouwen. Deze classificaties bestaan uit lineaire ondersteuningsvectormachine (SVC), Gaussiaanse naïeve Bayes (GNB), logistische regressieclassificatie (LR), k-dichtstbijzijnde buur, waarbij k standaard is ingesteld op 5 (KNN), XGBoost, willekeurig bos (RF) en beslissingsboom (DT).
De willekeurige splitsing van de dataset in de trein: testsubsets kunnen worden ingesteld in de opdrachtregel. In het gedemonstreerde voorbeeld wordt de verhouding trein: test = 8: 2 gebruikt.
