Il trascrittoma è costituito dall’espressione di tutti i geni in un campione e può essere profilato mediante tecnologie ad alto rendimento come il microarray e l’RNA-seq1. I livelli di espressione di un gene in un set di dati sono chiamati caratteristica trascrittomica e la rappresentazione differenziale di una caratteristica trascrittomica tra il fenotipo e i gruppi di controllo definisce questo gene come biomarcatore di questo fenotipo 2,3. I biomarcatori trascrittomici sono stati ampiamente utilizzati nelle indagini sulla diagnosi di malattia4, sul meccanismo biologico5 e sull’analisi di sopravvivenza 6,7, ecc.
I modelli di attività genica nei tessuti sani trasportano informazioni cruciali sulla vita 8,9. Questi modelli offrono informazioni preziose e fungono da riferimento ideali per comprendere le complesse traiettorie di sviluppo dei disturbi benigni 10,11 e delle malattie letali12. I geni interagiscono tra loro e i trascrittomi rappresentano i livelli di espressione finale dopo le loro complicate interazioni. Tali modelli sono formulati come rete di regolazione trascrizionale13 e rete metabolica14, ecc. L’espressione degli RNA messaggeri (mRNA) può essere regolata trascrizionalmente da fattori di trascrizione (TF) e lunghi RNA intergenici non codificanti (lincRNAs)15,16,17. L’analisi convenzionale dell’espressione differenziale ha ignorato tali complesse interazioni geniche con l’assunzione di indipendenza inter-caratteristica18,19.
I recenti progressi nelle reti neurali a grafo (GNN) dimostrano un potenziale straordinario nell’estrazione di informazioni importanti dai dati basati su OMIC per gli studi sul cancro20, ad esempio identificando i moduli di co-espressione21. La capacità innata dei GNN li rende ideali per modellare le intricate relazioni e dipendenze tra i geni22,23.
Gli studi biomedici spesso si concentrano sulla previsione accurata di un fenotipo rispetto al gruppo di controllo. Tali compiti sono comunemente formulati come classificazioni binarie 24,25,26. In questo caso, le due etichette di classe sono in genere codificate come 1 e 0, vero e falso o anche positivo e negativo27.
Questo studio mirava a fornire un protocollo di facile utilizzo per la generazione della vista di regolazione trascrizionale (mqTrans) di un set di dati di trascrittoma basato sul modello di riferimento GAT (graph-attention network) pre-addestrato. Il framework GAT multitasking di un lavoroprecedentemente pubblicato 26 è stato utilizzato per trasformare le caratteristiche trascrittomiche nelle caratteristiche mqTrans. Un ampio set di dati di trascrittomi sani della piattaforma Xena28 dell’Università della California, Santa Cruz (UCSC) è stato utilizzato per pre-addestrare il modello di riferimento (HealthModel), che ha misurato quantitativamente le regolazioni di trascrizione dai fattori regolatori (TF e lincRNA) agli mRNA bersaglio. La vista mqTrans generata potrebbe essere utilizzata per costruire modelli di previsione e rilevare biomarcatori oscuri. Questo protocollo utilizza il set di dati dei pazienti con adenocarcinoma del colon (COAD) dal database The Cancer Genome Atlas (TCGA)29 come esempio illustrativo. In questo contesto, i pazienti in stadio I o II sono classificati come campioni negativi, mentre quelli in stadio III o IV sono considerati campioni positivi. Vengono inoltre confrontate le distribuzioni dei biomarcatori oscuri e tradizionali nei 26 tipi di cancro TCGA.
Descrizione della pipeline HealthModel
La metodologia impiegata in questo protocollo si basa sul framework26 precedentemente pubblicato, come delineato nella Figura 1. Per iniziare, gli utenti devono preparare il set di dati di input, inserirlo nella pipeline HealthModel proposta e ottenere le funzionalità mqTrans. Le istruzioni dettagliate per la preparazione dei dati sono fornite nella sezione 2 della sezione relativa al protocollo. Successivamente, gli utenti hanno la possibilità di combinare le funzionalità mqTrans con le caratteristiche trascrittomiche originali o di procedere solo con le caratteristiche mqTrans generate. Il set di dati prodotto viene quindi sottoposto a un processo di selezione delle caratteristiche, con gli utenti che hanno la flessibilità di scegliere il valore preferito per k nella convalida incrociata k-fold per la classificazione. La metrica di valutazione principale utilizzata in questo protocollo è l’accuratezza.
HealthModel26 classifica le caratteristiche trascrittomiche in tre gruppi distinti: TF (fattore di trascrizione), lincRNA (RNA non codificante intergenico lungo) e mRNA (RNA messaggero). Le caratteristiche del TF sono definite in base alle annotazioni disponibili nell’Atlante delle Proteine Umane30,31. Questo lavoro utilizza le annotazioni dei lincRNA dal set di dati GTEx32. I geni appartenenti alle vie di terzo livello nel database KEGG33 sono considerati come caratteristiche dell’mRNA. Vale la pena notare che se una caratteristica dell’mRNA mostra ruoli regolatori per un gene bersaglio, come documentato nel database TRRUST34, viene riclassificata nella classe TF.
Questo protocollo genera anche manualmente i due file di esempio per gli ID dei geni dei fattori regolatori (regulatory_geneIDs.csv) e dell’mRNA bersaglio (target_geneIDs.csv). La matrice di distanza a coppie tra le caratteristiche regolatorie (TF e lincRNA) è calcolata dai coefficienti di correlazione di Pearson e raggruppata mediante la popolare analisi della rete di co-espressione genica pesata su strumenti (WGCNA)36 (adjacent_matrix.csv). Gli utenti possono utilizzare direttamente la pipeline HealthModel insieme a questi file di configurazione di esempio per generare la vista mqTrans di un set di dati trascrittomico.
Dettagli tecnici di HealthModel
HealthModel rappresenta le intricate relazioni tra TF e lincRNA come un grafo, con le caratteristiche di input che fungono da vertici indicati da V e una matrice di bordi inter-vertice designata come E. Ogni campione è caratterizzato da caratteristiche regolatorie K , simboleggiate da VK×1. In particolare, il set di dati comprendeva 425 TF e 375 lincRNA, risultando in una dimensionalità del campione di K = 425 + 375 = 800. Per stabilire la matrice dei bordi E, questo lavoro ha utilizzato il popolare strumento WGCNA35. Il peso a coppie che collega due vertici rappresentati come 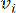 e , è determinato dal coefficiente
e , è determinato dal coefficiente 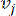 di correlazione di Pearson. La rete di regolazione genica presenta una topologia scale-free36, caratterizzata dalla presenza di geni hub con ruoli funzionali cardine. Calcoliamo la correlazione tra due caratteristiche o vertici, e , usando la misura di sovrapposizione topologica (TOM) come segue:
di correlazione di Pearson. La rete di regolazione genica presenta una topologia scale-free36, caratterizzata dalla presenza di geni hub con ruoli funzionali cardine. Calcoliamo la correlazione tra due caratteristiche o vertici, e , usando la misura di sovrapposizione topologica (TOM) come segue:
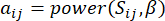 (1)
(1)
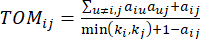 (2)
(2)
Il β di soglia soft viene calcolato utilizzando la funzione ‘pickSoft Threshold’ del pacchetto WGCNA. Viene applicata la funzione esponenziale di potenza aij , dove 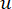 rappresenta un gene escludendo i e j, e
rappresenta un gene escludendo i e j, e 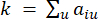 rappresenta la connettività dei vertici. WGCNA raggruppa i profili di espressione delle caratteristiche trascrittomiche in più moduli utilizzando una misura di dissimilarità comunemente impiegata (
rappresenta la connettività dei vertici. WGCNA raggruppa i profili di espressione delle caratteristiche trascrittomiche in più moduli utilizzando una misura di dissimilarità comunemente impiegata (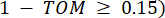 37.
37.
Il framework HealthModel è stato originariamente progettato come un’architettura di apprendimento multitasking26. Questo protocollo utilizza solo l’attività di pre-addestramento del modello per la costruzione della vista mqTrans trascrittomica. L’utente può scegliere di perfezionare ulteriormente l’HealthModel pre-addestrato nell’ambito della rete di attenzione del grafico multitask con ulteriori campioni trascrittomici specifici dell’attività.
Dettagli tecnici della selezione e della classificazione delle funzionalità
Il pool di selezione delle funzionalità implementa undici algoritmi di selezione delle funzionalità (FS). Tra questi, tre sono algoritmi FS basati su filtri: la selezione delle migliori caratteristiche K utilizzando il coefficiente di informazione massima (SK_mic), la selezione delle caratteristiche K in base all’FPR della MIC (SK_fpr) e la selezione delle caratteristiche K con il più alto tasso di falsa scoperta della MIC (SK_fdr). Inoltre, tre algoritmi FS basati su alberi valutano le singole funzionalità utilizzando un albero decisionale con l’indice di Gini (DT_gini), gli alberi decisionali potenziati adattivi (AdaBoost) e la foresta casuale (RF_fs). Il pool incorpora anche due metodi wrapper: l’eliminazione ricorsiva delle funzionalità con il classificatore del vettore di supporto lineare (RFE_SVC) e l’eliminazione ricorsiva delle funzionalità con il classificatore di regressione logistica (RFE_LR). Infine, sono inclusi due algoritmi di incorporamento: classificatore SVC lineare con i valori di importanza delle funzionalità L1 (lSVC_L1) più alti e classificatore di regressione logistica con i valori di importanza delle funzionalità L1 (LR_L1).
Il pool di classificatori utilizza sette classificatori diversi per compilare modelli di classificazione. Questi classificatori comprendono la macchina a vettori di supporto lineare (SVC), la naïve Bayes gaussiana (GNB), il classificatore di regressione logistica (LR), il vicino più vicino k, con k impostato su 5 per impostazione predefinita (KNN), XGBoost, foresta casuale (RF) e albero decisionale (DT).
La suddivisione casuale del set di dati nei sottoinsiemi di test train: può essere impostata nella riga di comando. Nell’esempio illustrato viene utilizzato il rapporto train: test = 8: 2.
