Fonte: Jonathan F. Blaize1, Elizabeth Suter1e Christopher P. Corbo1
1 Dipartimento di Scienze Biologiche, Wagner College, 1 Campus Road, Staten Island NY, 10301
La valutazione quantitativa dei procarioti può essere onerosa data la loro abbondanza, la propensione alla proliferazione esponenziale, la diversità delle specie all’interno di una popolazione e le specifiche esigenze fisiologiche. Ad aggravare questa sfida, è la natura a quattro fasi in cui i batteri si replicano (ritardo, log, stazionario e morte). La capacità di stimare con precisione la concentrazione di microrganismi è necessaria per il successo dell’identificazione, dell’isolamento, della coltivazione e della caratterizzazione (6). Pertanto, i microbiologi hanno impiegato la diluizione seriale e varie tecniche di placcatura per oltre un secolo per quantificare in modo affidabile la carica batterica e virale in ambienti di laboratorio clinici, industriali, farmaceutici e accademici (2,4,6). Le descrizioni di questa metodologia apparvero per la prima volta nel 1883 quando lo scienziato e medico tedesco Robert Koch pubblicò il suo lavoro sugli agenti infettivi che causano malattie (2). Spesso indicato come il padre della batteriologia moderna, le tecniche prenominate di Koch sono diventate il gold standard per l’enumerazione di microrganismi, coltivabili o meno, in tutto il mondo.
La diluizione seriale è una riduzione sistematica di un’entità nota o sconosciuta (un soluto, un organismo, ecc.) attraverso la successiva risos sospensione di una soluzione iniziale(soluzione 0) in volumi fissi di un diluente liquido (spazi vuoti). Questi spazi vuoti di solito consistono di 0,45% di soluzione salina, anche se la composizione può essere variata (7). Mentre uno sperimentatore può scegliere qualsiasi volume per ogni diluente, è più spesso un multiplo di 10, facilitando la riduzione logaritmica del campione. Ad esempio, lasoluzione 0 contiene un totale di 100 cellule di E. coli sospese in 10 ml di brodo nutritivo. Se 1 mL disoluzione 0 viene rimosso e aggiunto a 9 mL di soluzione salina (diluente1), la nuovasoluzione (soluzione 1) conterrebbe 1/10 della concentrazione iniziale di E. coli. In questo esempio, la nuova soluzione(soluzione 1) conterrebbe 10 cellule di E. coli. Ripetendo questo processo rimuovendo 1 mL disoluzione 1 e aggiungendolo ad altri 9 mL di soluzione salina (diluente2)si produrrebbela soluzione 2, contenente solo una singola cellula di E. coli. Poiché ogni nuova soluzione (9 mL di diluente + 1 mL di soluzione) contiene un totale di 10mL, possiamo concludere che il fattore di diluizione per questa riduzione è 10 o che si è tratta di una diluizione seriale 10 volte (Figura 1). Poiché abbiamo iniziato solo con 100 cellule in questo esempio e stiamo diluendo di un fattore 10, sono necessari solo due passaggi per raggiungere la concentrazione minima assoluta di 1 cellula.
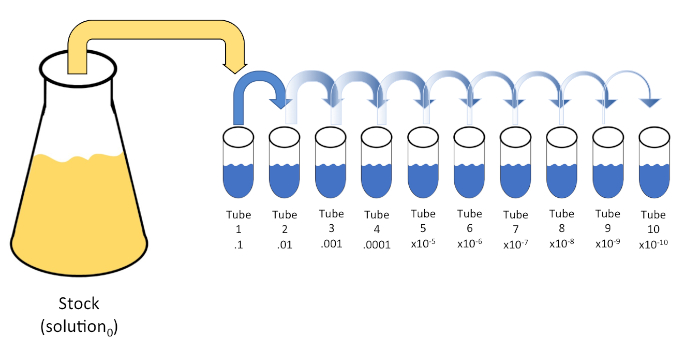
Figura 1: Diluizione seriale di una soluzione stock. Un’aliquota di 1 mL della soluzione stock(soluzione 0) viene aggiunta al tubo 1 che contiene 9 mL di soluzione salina allo 0,45% (dilent1); il prodotto di questa miscela è lasoluzione 1. Ripetere aliquotando 1 mL dellasoluzione appena creata 1 e aggiungendola al tubo 2. L’aliquotazione e la ricaspensione continuano in questo modo fino al raggiungimento del tubo finale, diluendo la concentrazione di stock di un fattore di 10 ciascuno ad ogni passo. Fare clic qui per visualizzare una versione più grande di questa figura.
La diluizione seriale è la tecnica più semplice per ottenere concentrazioni gestibili di un organismo desiderato ed è completata da striature e spalmature di Petri, solo due delle molte tecniche di placcatura utilizzate dai microbiologi. Questo vantaggio di questo approccio è che lo sperimentatore può raccogliere ceppi puri di una singola specie o ceppi separati da una popolazione mista (7). La striatura si ottiene introducendo un organismo in un mezzo solido (generalmente costituito da agarose) su cui crescerà se sono disponibili i nutrienti appropriati. Spazzare delicatamente un ciclo inoculante sterile attraverso il mezzo (in modo che rimanga una striscia sottile) in un modello sinusoidale rigido distribuirà l’organismo proporzionalmente alla frequenza della forma d’onda dello sperimentatore. Dividere la capsula di Petri in terzi o quarti (striscia di quadrante) e diminuire la frequenza di ogni striscia quando viene inserita una nuova regione del piatto ridurrà gradualmente il numero di microrganismi che possono occupare quella regione, producendo singole colonie invece di un prato batterico non quantificabile. La placcatura diffusa non diluisce ulteriormente i campioni; uno spandi vetro sterile viene utilizzato per distribuire un’aliquota di mezzi di sospensione su un’intera capsula di Petri (Figura 2). Le colonie che crescono sulla piastra diffusa derivano da una singola cellula e ogni colonia sul piatto può essere contata per stimare il numero di unità formanti colonie per millilitro (CFU) in una data sospensione, rappresentata come CFU/mL (6) (Figura 3) L’agar morbido e la replica placcatura sono variazioni delle tecniche di cui sopra e consentono l’isolamento del batteriofago e lo screening mutante, rispettivamente (1,7).
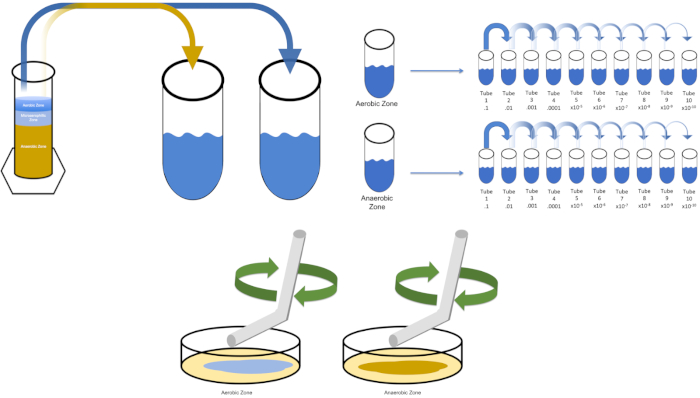
Figura 2: Striature di piastre per l’enumerazione batterica e l’isolamento del ceppo. Etichettare il fondo di una capsula di Petri con informazioni di identificazione (nome dei batteri, data, supporto) e dividere in terzi. Dopo aver selezionato una diluizione appropriata del campione di riserva, prendere un anello di inoculazione sterile (monouso o fiammato) e immergerlo nella provetta (qui, T3). Sollevare leggermente il coperchio della capsula di Petri su un lato in modo che solo l’anello inoculante possa accedere all’agar. Fai scivolare il loop inoculante attraverso la parte superiore del supporto a zig-zag facendo attenzione a non compromettere l’agar. Ruotare la piastra di circa 1/3 (~118°) e ridurre la frequenza del movimento a zig-zag. Ruota un ultimo tempo e riduci ancora una volta la frequenza a zig-zag. Fare clic qui per visualizzare una versione più grande di questa figura.
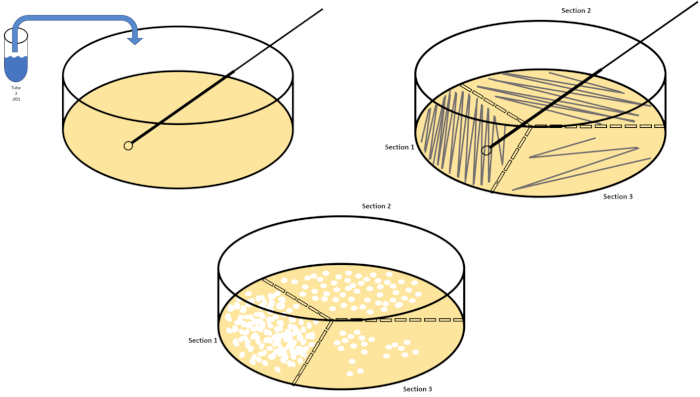
Figura 3: Placcatura diffusa. 1 g della zona aerobica è stato sospenso in T1 e poi diluito in serie. Un’asta sterile di spargimento monouso in vetro o plastica viene utilizzata per distribuire l’inoculo in ogni piatto. Questo è stato ripetuto con 1 g della zona anaerobica. Fare clic qui per visualizzare una versione più grande di questa figura.
Come per le diluizioni seriali, viene utilizzata una scala logaritmica per esprimere la concentrazione dell’organismo. Il numero di colonie coltivate in piastre di Petri standard di 100 mm x15 mm può essere enumerato manualmente (o automatizzato con l’aiuto dell’elaborazione computazionale) identificando cluster isolati di crescita. I conteggi che ammontano a meno di 30 o superiore a 300 devono essere definiti rispettivamente come troppo pochi da contare (TFTC) o troppo numerosi da contare (TNTC). Nel caso di quest’ultimo, deve essere eseguita una diluizione seriale per ridurre la concentrazione prima di riaffermire una nuova capsula di Petri. Facendo la media del numero di colonie autonome identificate da tre piastre di Petri separate e moltiplicando la media per il fattore di diluizione si otterrà CFU/mL; tracciare il log10 di CFU/mL contro il tempo rivelerà il tempo medio di generazione dell’organismo (7).