Fuente: Tilde Andersson1, Rolf Lood1
1 Departamento de Ciencias Clínicas Lund, División de Medicina de Infecciones, Centro Biomédico, Universidad de Lund, 221 00 Lund, Suecia
Los virus que infectan los organismos procaloicos, llamados bacteriófagos o simplemente fagos, fueron identificados a principiosdel siglo XX por Twort (1) y d’Hérelle (2) de forma independiente. Desde entonces, los fagos han sido ampliamente reconocidos por su valor terapéutico (3) y su influencia en los ecosistemas humanos (4), así como en los ecosistemas globales (5). Las preocupaciones actuales han impulsado un renovado interés en el uso de fagos como alternativa a los antibióticos modernos en el tratamiento de enfermedades infecciosas (6). Esencialmente toda la investigación de fagose se basa en la capacidad de purificar y cuantificar virus, también conocido como un titer viral. Descrito inicialmente en 1952, este fue el propósito del ensayo de placa (7). Décadas y múltiples avances tecnológicos más tarde, el ensayo de placa sigue siendo uno de los métodos más fiables para la determinación del titer viral (8).
Los bacteriófagos subsisten inyectando su material genético en las células huésped, secuestrando las máquinas para la producción de nuevas partículas de fago, y eventualmente causando que el huésped libere numerosos viriones de progenie a través de la lisis celular. Debido a su tamaño minúsmico, los bacteriófagos no se pueden observar utilizando únicamente microscopía de luz; por lo tanto, se requiere microscopía electrónica de barrido (Figura 1). Además, los fagos no se pueden cultivar en placas de agar nutricionales como bacterias, ya que necesitan células huésped para abeste.
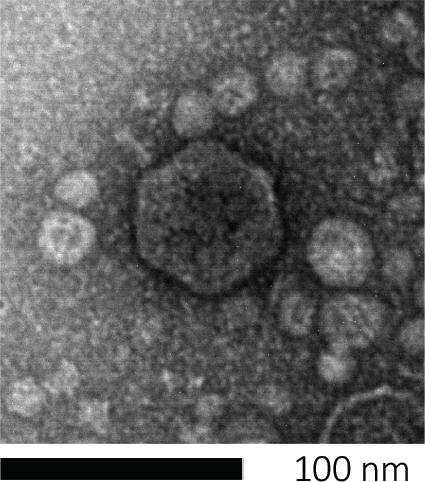
Figura 1: La morfología de un bacteriófago, aquí ejemplificada por un fago E. coli, se puede estudiar mediante microscopía electrónica de barrido. La mayoría de los bacteriófagos pertenecen a Caudovirales (bacteriófagos de cola). Este fago en particular tiene una estructura de cola muy corta y una cabeza icosahedral, colocándola en la familia de Podovirus.
El ensayo de placa (Figura 2) se basa en la incorporación de células huésped, preferentemente en el crecimiento de la fase log, en el medio. Esto crea una capa densa y turbia de bacterias capaces de mantener el crecimiento viral. Un fago aislado puede infectar, replicar y oxidar una célula. Con cada célula lysed, varias de las adyacentes se infectan inmediatamente. Varios ciclos en, una zona clara (una placa) se pueden observar en la placa de otro modo turbia (Figura 2B/Figura 3A),lo que indica la presencia de lo que inicialmente era una sola partícula de bacteriófago. Por lo tanto, el número de unidades formadoras de placas por volumen(es decir, PFU/ml) de una muestra puede determinarse a partir del número de placas generadas.
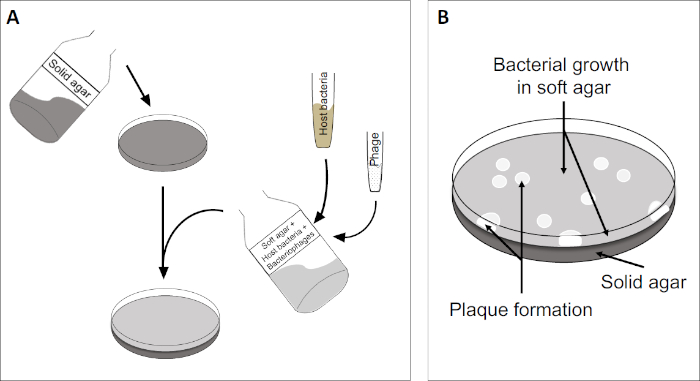
Figura 2: Las pruebas para las unidades formadoras de placas (PFU) son un método común para determinar el número de bacteriófagos en una muestra. (A) La base de una placa estéril de Petri está cubierta con un medio de nutrientes sólido adecuado, seguido de una mezcla de medios blandos, células huésped susceptibles y una dilución de la muestra original de bacteriófagos. Tenga en cuenta que la suspensión del fago podría, en algunos casos, también extenderse uniformemente a través de la superficie del agar blando ya solidificado. (B) El crecimiento de las bacterias anfitrionas forma un césped de células en la capa superior de agar. La replicación de bacteriófagos genera zonas claras, o placas, causadas por la lisis de la célula huésped.
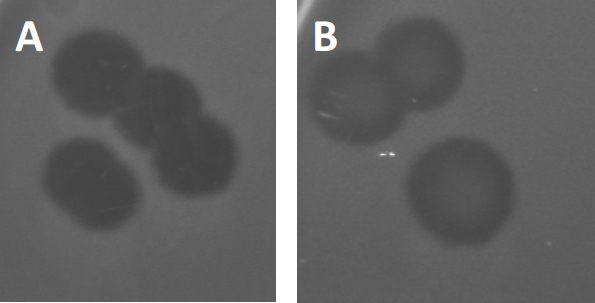
Figura 3: Los resultados de las pruebas de PFU muestran múltiples placas generadas por bacteriófagos. Debido a la lisis de las células huésped susceptibles, las placas pueden ser vistas como zonas de despeje en el césped bacteriano, ya sea con (A) aclaramiento completo, o (B) re-crecimiento parcial causado por la generación de bacterias resistentes (o posiblemente por fagos templados en el lisogénico).
Ciertos fagos templados pueden adoptar lo que se conoce como un ciclo de vida lisogénico, además del crecimiento lítico descrito anteriormente. En la lisógena, el virus asume un estado latente mediante la incorporación de su material genético en el genoma de la célula huésped (9), a menudo confiriendo resistencia a más infecciones por fagos. Esto a veces se revela a través de una ligera opacidad de la placa (Figura 3B). Vale la pena señalar sin embargo, que las placas también pueden aparecer borrosas debido al re-crecimiento de bacterias que han evolucionado resistencia al fago independiente de las infecciones anteriores de fago.
Los virus pueden adjuntarse, o adsorgar, a sólo un rango limitado de bacterias anfitrionas (10). Los rangos de acogida están aún más limitados por estrategias antivirales intracelulares como el sistema CRISPR-Cas (11). La resistencia/sensibilidad hacia fagos específicos mostrados por subgrupos bacterianos se ha utilizado históricamente para clasificar las cepas bacterianas en diferentes tipos de fagos (Figura 4). Aunque la eficacia de este método ha sido ahora superada por nuevas técnicas de secuenciación, la tipificación de fagos todavía puede proporcionar información valiosa sobre las interacciones entre bacterias y fagos, por ejemplo, facilitando el diseño de un cóctel de fago para uso clínico .
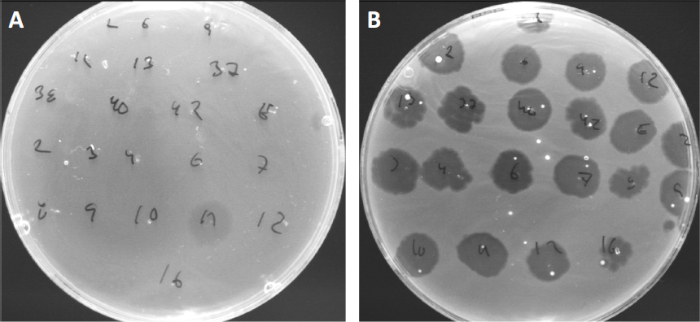
Figura 4: Sensibilidad del fago de diferentes cepas bacterianas. Las placas de agar blandas con cepa Cutibacterium acnes (A) AD27 y (B) AD35, fueron manchadas con 21 bacteriófagos Diferentes de C. acnes. Sólo el fago 11 fue capaz de infectar y matar AD27 mientras que la cepa AD35 mostró sensibilidad hacia todos los fagos. Esta técnica, que se denomina tipificación de fagos, se puede utilizar para dividir especies bacterianas y cepas en diferentes subgrupos basados en la susceptibilidad del fago.