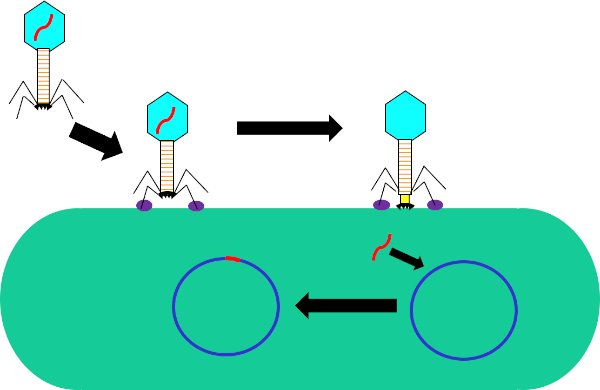
Bacteria can adapt quickly to a fast-changing environment by exchanging genetic material and one way they can do this is via transduction, the exchange of genetic material mediated by bacterial viruses. A bacteriophage, often abbreviated to phage, is a type of virus that infects bacteria by first attaching to the surface of the host and then injecting its DNA into the bacterial cell. It then degrades the host cell’s own DNA and replicates its viral genome, whilst hijacking the cell’s machinery to synthesize many copies of its proteins. These phage proteins then self-assemble and package the phage genomes to form multiple progeny. However, due to the low fidelity of the DNA packaging mechanism, occasionally, the phage packages fragments of bacterial DNA into the phage capsid. After inducing the lysis of the host, the phage progeny are released and, once such a phage infects another host cell, it transfers the DNA fragment of its previous host. This can then recombine and become permanently incorporated into the new host’s chromosome, thereby mediating gene transfer between the two bacteria.
To carry out phage transduction in the laboratory requires a donor strain that contains a gene of interest, a recipient strain that lacks it, a phage that can infect both the strains, and a method to select the transduced bacteria. In most cases, this will be a selective solid growth media that supports the growth of transduced bacteria but inhibits the growth of non-transduced ones. To begin, the donor strain that contains the gene of interest is cultured in a liquid growth medium. When all the bacteria are actively dividing in the log phase of their growth, the culture is inoculated with the target phage. After three to four hours of incubation, when nearly all the bacteria have lysed and released the phage particles, the donor phage lysate is inoculated into a freshly grown culture of the recipient bacterial strain. After a brief incubation of one hour, the culture should now contain a mixture of transduced and non-transduced bacterial cells and this is screened for the transduced cells by spreading a fraction of the suspension onto an appropriate selective solid growth media. Upon further incubation, the transduced cells should grow and multiply to yield visible colonies. These colonies can then be selected for further analysis using a variety of methods to further confirm successful transduction, such as colony PCR, DNA sequencing, or quantitative PCR.
Before starting the procedure, put on any appropriate personal protective equipment, including a lab coat and gloves. Next, sterilize the workspace with 70% ethanol and wipe down the surface.
After this, prepare three one-milliliter aliquots of LB salt solution. Now, prepare a donor strain culture by adding 100 microliters of E. coli to a 15 milliliter conical vial containing five milliliters of LB growth medium with 500 micrograms of ampicillin. Then, grow the culture overnight at 37 degrees Celsius with aeration and shaking at 220 rpm. The next day, wipe down the bench top with 70% ethanol before removing the culture from the shaking incubator. Next, dilute the overnight culture one to 100 by adding 10 microliters of donor strain to 990 microliters of fresh LB supplemented with salt solution.
Allow the bacterial dilution to grow at 37 degrees Celsius for two hours with aeration and shaking at 220 rpm. Once the cells have reached early log phase, remove the culture from the incubator, add 40 microliters of P1 phage to the culture and incubate again. Continue to monitor the cells for one to three hours until the culture has lysed. Next, add 50 to 100 microliters of chloroform to the lysate and mix by vortexing. Then, centrifuge the lysate to remove debris and transfer the supernatant to a fresh tube. Add a few drops of chloroform to the supernatant and store it at four degrees Celsius for no more than one day.
To begin the transduction procedure, obtain a one milliliter culture of recipient strain. Next, transfer 100 microliters of donor phage lysate into a 1.5 milliliter microcentrifuge tube and incubate it at 37 degrees Celsius with the cap open for 30 minutes to allow any remaining chloroform to evaporate. While the donor phage lysate incubates, pellet the recipient strain cells via gentle centrifugation. Discard the supernatant and resuspend the cell pellet in 300 microliters of fresh LB containing 100 millimolar magnesium sulfate and five millimolar calcium chloride.
Next, set up the transduction reaction by combining 100 microliters of the recipient strain and 100 microliters of the donor phage lysate in a microcentrifuge tube. Then, set up the negative control by combining 100 microliters of the recipient strain and 100 microliters of the LB with magnesium sulfate and calcium chloride. After incubation, add 200 microliters of one molar sodium citrate and one milliliter of LB to both tubes, and mix by gently pipetting up and down. Then, after the tubes have been incubated for an hour, gently pellet the cells via centrifugation.
After centrifuging, discard the supernatant and resuspend the pelleted cells in 100 microliters of LB with 100 millimolar sodium citrate. Vortex the solutions and pipette the entire transduced sample onto an LB agar plate with 1X ampicillin. Finally, pipette the entire volume of the negative control cell mixture onto an LB agar plate without ampicillin. After incubating the plates overnight at 37 degrees Celsius, use a sterile pipette tip to pick three to four colonies from the transduction plate and streak them onto a new LB agar plate containing 1X ampicillin and 100 microliters of one molar sodium citrate. Repeat this plating method for the negative control on another LB agar plate containing only 100 microliters of one molar sodium citrate. Then, incubate the plates at 37 degrees Celsius overnight to allow colonies free of phage to grow.
The next day, wipe down the bench top with 70% ethanol before removing your plates from the incubator. Using a sterile pipette tip, pick three colonies from the transduction plate and add them each to a separate tube containing five milliliters of LB media. Then, select three colonies from the negative control plate and add them to another tube containing five milliliters of LB media. Grow the cultures overnight at 37 degrees Celsius with aeration and shaking at 220 rpm. After sterilizing the bench top as previously demonstrated, use a DNA miniprep kit to isolate DNA from 4.5 milliliters of each culture according to the manufacturer’s instructions. Then, elute the DNA with 35 microliters of nuclease-free water and measure the resulting concentration by lab spectrophotometer. Finally, prepare glycerol stocks by adding the remaining 0.5 milliliters of both bacterial solutions to 0.5 milliliters of 100% glycerol.
To confirm transduction, first prepare two qPCR master mixes for 24 qPCR reactions. For the first master mix, add 150 microliters of qPCR buffer mix to a microcentrifuge tube and 12 microliters each of a forward and reverse primer designed to amplify the ampicillin resistance gene. Next, prepare a second qPCR master mix by adding 150 microliters of qPCR master mix to a microcentrifuge tube and then adding 12 microliters each of a forward primer and reverse primer designed to amplify a housekeeping gene.
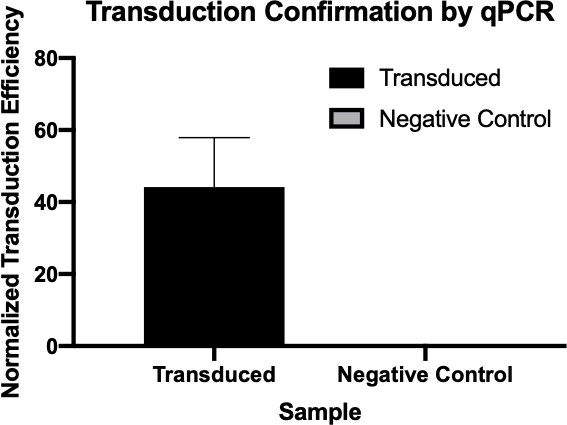
For each qPCR reaction, combine 100 micrograms of experimental DNA from each reaction with 14.5 microliters of qPCR master mix. Now, prepare the remaining reactions as previously demonstrated. Transfer the reactions to a thermocycler preheated to 94 degrees Celsius and then initiate the program. Finally, use the cycle quantification, or Cq, values generated by qPCR to calculate the normalized transduction efficiency of the ampicillin resistance gene.
The cycle quantitation, or Cq, values for the genes of interest were tabulated for each of the negative controls and transduced samples. Low Cq values, typically below 29 cycles, like the transduced samples in this example indicate high amounts of the target sequence.
A housekeeping gene, also tabulated here, is used as a loading control to normalize the amount of DNA in each reaction and as a positive control to ensure the qPCR is working. Provided the same amounts of the housekeeping gene are loaded, it is found at relatively the same rate in each sample.
Next, to calculate the delta Cq value for each sample, subtract the Cq value of the housekeeping gene for each sample from the Cq value of its corresponding target gene. For example, the delta Cq of the first negative control is 13.54. Then, use this value to calculate the normalized transduction efficiency of each sample using the formula shown here. Finally, the average normalized transduction efficiency for each sample group can be calculated.