An enriched population of astrocytes was acquired via FACS sorting of eGFP-S100β transgenic animals 27. Due to decreasing quality of cells and molecular molecules isolated from animals older than postnatal day 50 (p50), animals aged p0-p40 are optimal for such experiments. Cortical tissue was used for these experiments. Cortices from two to six animals were pooled together. FACS was performed at UAB Comprehensive Flow Cytometry Core facility. Sorting was performed on Becton Dickinson FacsAria II. eGFP excitation was obtained using 488 nm laser; no compensation was necessary. A gated population was targeted based on forward and side scatter (Figure 1B). A live cell population was determined using an ethidium bromide dead cell indicator and gated (Figure 1C). Two different populations were observed based on the eGFP profile (Figure 1D). Through empiric testing, the cell population with higher eGFP profile was identified as the eGFP-positive cell population (Figure 1F-F''). Visual analysis of the second population with a lower eGFP profile revealed cell fragments expressing eGFP, but devoid of nuclei, likely representing debris from astrocytic processes (Figure 1G-G''). Representative images of dissociated eGFP positive astrocytes following dissociation but prior to sorting (Figure 2A) and after sorting (Figure 2B) are shown. Isolated eGFP positive cell population demonstrated a 40-fold increase in ALDH1L1 mRNA, an astrocytic specific marker 39 (Figure 2C). Additionally, despite shared expression of S100β in NG2+ OPCs and astrocytes 39, we observed a 4-fold reduction of NG2 mRNA, a marker for oligodendrocyte precursor cells 39, indicating isolation of an enriched astrocytic cell population (Figure 2C-D). RNA and DNA isolated from FACS sorted astrocytes were of sufficient quality and quantity to be used for subsequent methylation studies. Total RNA and DNA isolated from varying ages and number of animals is listed as a reference for expected yield of molecular molecules following FACS (Table 5). It should be noted that the yield will also vary based on number of animals utilized, incubation period, and experience of researcher. Table 6 demonstrates the variability of events acquired depending on number of animals utilized and incubation periods.
For methylation studies, starting with high quality and sufficient quantities of DNA is essential for successful downstream applications. To ensure sufficient lysis of tissue for RNA or DNA extraction, all homogenization of tissue or cells was followed by trituration using a 23G needle 7x, taking care to avoid frothing during lysis. Once high quality DNA is extracted and bisulfite converted, regions of interest can be targeted for MS-HRMA. Note that when assessing quality and concentration of bisulfite converted DNA, spectrophotometer should be set to read at an OD factor of 33 as bisulfite converted DNA is single stranded. The 260/280 values for bisulfite converted DNA ranged from 2.20-2.70.
Before beginning methylation studies, it is critical to appropriately select and calibrate a thermal cycler for MS-HRMA studies. Additionally, one must determine how to export and utilize raw data generated from MS-HRMA. Applied Biosystems High Resolution Melting Getting Started Guide was utilized to assist in calibrating the 7900HT thermal cycler. Finally, data was extracted and imported into HRM analysis software for subsequent data analysis. As detailed in the protocol, pre and post start and stop parameters were set near the transitions of the melt curve. Peak temperature differences were used to extrapolate methylation status of unknown samples. Estimated methylation data acquired from MS-HRMA corresponded closely to methylation data generated from pyrosequencing (pyrosequencing data was generated in house using primers targeting same regions as MS-HRM primers) (Figure 3C) 15.
A dual luciferase assay was utilized to assess the transcriptional activity of hyper-methylated regions of the gene of interest (Figure 4A). Each CpG-island-luc2 plasmid was linearized and then methylated. However, because both the insert (CpG island) and vector (luc2) are methylated during the reaction, one must excise the methylated insert and re-ligate to a non-methylated vector. Due to extensive manipulation of the plasmid, significant losses occur and one must begin with sufficient starting material. HpaII digestion was utilized to verify the methylation status of each plasmid as HpaII only digests non-methylated DNA (Figure 4B). It should be noted that if O/N methylation is insufficient, an additional 1x of SAM and 1x of CpG methylase may be added following 1 hr of incubation at 30 °C. Continue O/N incubation following addition of supplemental SAM and CpG methylase. Generation of methylated and non-methylated CpG-island-luc2 plasmid allows for direct assessment of changes between DNA methylation and transcriptional activity via measurement of luciferase activity.
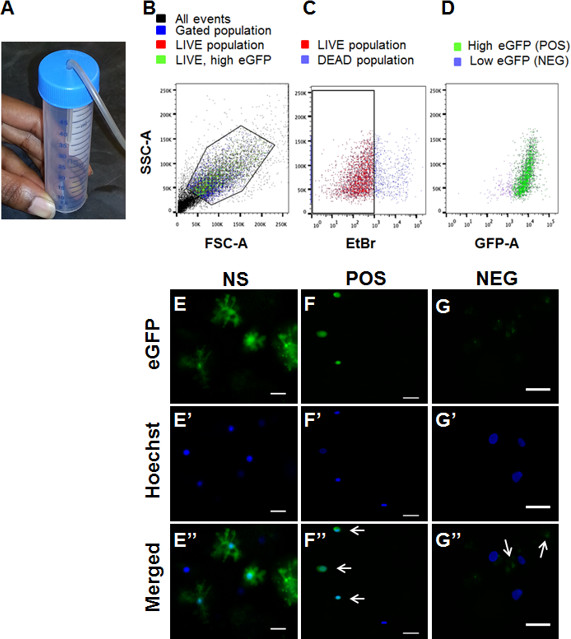
Figure 1. FACS sorting isolates eGFP+ cell popoulation. (A) Use of a 50 ml conical tube with hole cut on the top allows for surface gas exchange during papain digestion. (B) Sorted cells were gated based on forward and side scatter plots. (C) An ethidium bromide based dead cell indicator was utilized to gate a live cell population (red). (D) Live cell population demonstrated two populations – one with a high eGFP profile (green) and a low eGFP profile (purple). For all graphs, y-axis represents side scatter area (SSC-A); x-axis represents forward side scatter area (FSC-A), green fluorescent protein area (GFP-A), or ethidium bromide (EtBr). (E-G) Dissociated cells were incubated with Hoechst and 20 µl of dissociated cells was placed on coverslip and imaged. (E-E'') eGFP+ astrocytes, with extensive processes were visualized before sorting (NS). (F-F'') Following sorting, high eGFP+ population (POS) demonstrate eGFP+ cell soma which co-localize with Hoechst staining. (G-G'') Low eGFP population demonstrates eGFP+ staining which did not co-localize with Hoechst staining, likely representing astrocytic debris (for all images, scale = 20 µm). Please click here to view a larger version of this figure.
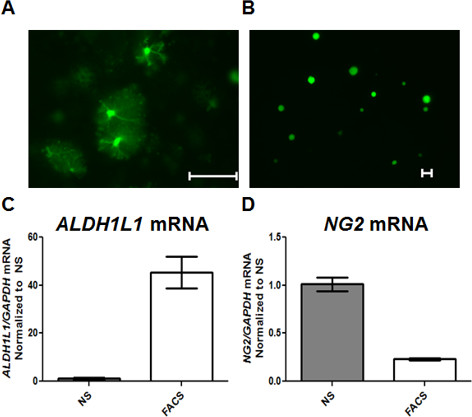
Figure 2. FACS sorting of eGFP-S100β transgenic animals provides enriched population of astrocytes. (A) 20 µl of dissociated cells before FACS (NS) are shown, processes remain intact (scale =50 µm). (B) Representative image post-FACS (FACS) cells are shown; astrocytic processes are removed and cell soma remains (scale =20 µm). (C) qPCR data demonstrates a 40-fold increase in ALDH1L1 mRNA in FACS sorted cells (FACS) compared to not sorted population (NS). (D) NG2 mRNA is reduced by 4-fold in FACS sorted cells compared to not sorted population (NS). Please click here to view a larger version of this figure.
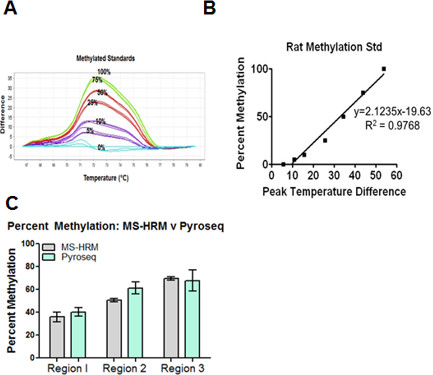
Figure 3. Sample data and analysis for MS-HRM studies. (A) Example of temperature difference plots generated from methylation standards. Corresponding percent methylation is labelled for each curve (B) Example of linear regression equation generated from rat methylated standards. (C) Comparison of the percent methylation acquired from pyrosequencing (pyroseq, green bars) and MS-HRMA (gray bars) demonstrates similar levels of methylation for varying regions of KCNJ10. Pyrosequencing data generated in house using primers targeting same regions as MS-HRM analysis. Please click here to view a larger version of this figure.
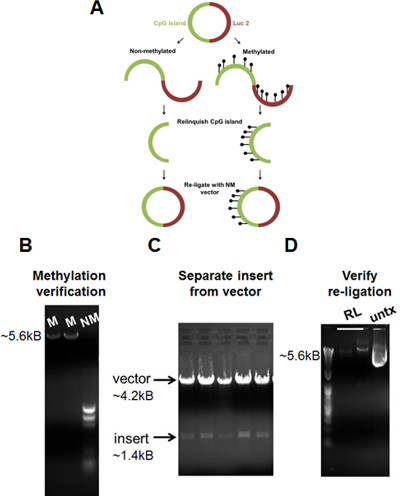
Figure 4. Dual luciferase assay allows for assessment of transcriptional activity of differentially methylated gene regions. (A) Diagram of luciferase transcriptional activity assay demonstrates workflow of protocol. (B) DNA gel displays differences in methylated versus non-methylated plasmids following HpaII digestion. Non-methylated DNA is readily digested following HpaII digestion compared to methylated DNA, demonstrating successful methylation of DNA plasmid. (C) DNA gel demonstrates separation of vector from insert following double digestion. Following DNA separation, vector and insert are gel extracted. (D) DNA gel demonstrates successful re-ligation of vector and insert. Re-ligated plasmid possesses similar molecular weight as untreated plasmid (untx). NM, non-methylated; M, methylated; RL, re-ligated; untx, untreated. Please click here to view a larger version of this figure.
| . | Volume (µl) | x3 + 10% excess | x # of samples |
| MeltDoc MM | 10 µl | 33 µl | |
| Primer 1 | 1.2 µl | 3.96 µl | |
| Primer 2 | 1.2 µl | 3.96 µl | |
| gDNA | 1 µl | — | |
| dH20 | 6.6 µl | 21.78 µl |
Table 1. Sample calculation of MeltDoctor Mastermix. Run each MS-HRM reaction in triplicate with 10% excess to compensate for pipetting error. Last column indicates calculation needed for multiple numbers of samples. Total volume for each reaction is 20 µl.
| Cycle | Temp (°C) | Time (sec) | Ramp rate (%) |
| 95 °C | :15 | 100 | |
| 40x | 95 °C | :15 | 100 |
| 60 °C | :60 | 100 | |
| Melting parameters | 95 °C | :10 | 100 |
| 60 °C | :60 | 100 | |
| 95 °C | :15 | 1 | |
| 60 °C | :15 | 100 | |
| Hold | 4 °C |
Table 2: Sample protocol for amplification. Denature samples at 95 °C for 10 min followed by 40 cycles of amplification and generation of melt curve. Melting parameters are listed within table. Note change in ramp rate that allows for acquisition of high- resolution melting temperature.
| Percent Methylation (Rat Standard) | Peak Temperature Difference |
| y | x |
| 0 | 5.670116 |
| 5 | 10.70448 |
| 10 | 15.5044 |
| 25 | 25.52295 |
| 50 | 34.43047 |
| 75 | 43.80894 |
| 100 | 53.88717 |
Table 3. Example of peak temperature difference data. Percent methylation standard is labelled as y-coordinate. Peak temperature difference data is labelled as x-coordinate. Representative peak temperature difference data was acquired using MS-HRM from one gene region.
| Peak Temp Difference | Estimated methylation |
| X | Y |
| Sample 1 (n=4) | |
| 47.450 | 81.115 |
| 46.292 | 78.657 |
| 41.694 | 68.894 |
| 47.292 | 80.779 |
| Sample 2 (n=4) | |
| 29.925 | 43.904 |
| 24.269 | 31.894 |
| 25.061 | 33.575 |
| 25.389 | 34.272 |
Table 4. Example of calculated estimated percent methylation. Peak temperature difference of samples of unknown methylation status is used to estimate percent methylation using a linear regression equation. Representative data from two different conditions, Sample 1 and Sample 2, are shown; n=4 for each condition.
| Age | Number of animals | Total RNA (ng) | Total DNA (ng) |
| p4-p10 | 3-6 | 455-868 | 265-1523 |
| p19-p22 | 1-2 | 220-680 | 583-1483 |
| p40-p50 | 3-4 | 198-260 | 578-1700 |
Table 5. Total RNA and DNA acquired from FACS sorted cells. Age and number of animals used in FACS experiments are listed. Note, both cortices were utilized for each N. Total RNA and DNA is listed as a reference for expected yield.
| Age | Number of animals | Pos Events | Neg Events | Incubation time (at 37 °C) |
| p26 | 1 | 2.18X10^6 | 1.15X10^6 | 20:00 min |
| p26 | 2 | 4.4X10^6 | 2.78X10^6 | 20:00 min |
| p26 | 2 | 9.2X10^6 | 3.3X10^6 | 30:00 min |
Table 6. Number of events acquired from FACS at postnatal day 26. At p26 number of positive (eGFP+) events and negative events varies depending on number of animals utilized and incubation period.
