1. Transgenic and Mutant Lines
- Establish/acquire transgenic zebrafish lines that have cell types of interest differentially labeled with fluorescent reporter variants. Optimally, the excitation/emission profile of the selected reporters should have minimal overlap (e.g., ECFP and EYFP), however this is not absolutely required. For our purposes, the use of membrane tethered fluorescent reporters greatly facilitates imaging of fine cellular details, such as neuritic processes, to resolve individual cell numbers and/or morphologies in large comingled groups and to ‘highlight’ regions of high membrane content, such as synaptic neuropils.
- For imaging at late larval stages, it is helpful to derive/cross transgenic lines into mutant backgrounds which reduce pigmentation [e.g., roy orbison (roy) or roy orbison; albino (roy;alb); 4]. In our experience, reduction of iridiphores (e.g., roy) is the most critical issue, melanogenesis can be addressed chemically using 1-phenyl 2-thiourea (PTU) or with “albino” mutants which lack melanophores. Note however that Ren et al.4, have demonstrated visual deficits and modest morphological changes in retinas of fish lacking melanaphores, and that intense light can induce photoreceptor death in “albino” mutants. Thus, these issues need to be taken into consideration when designing imaging experiments and/or interpreting data.
- For this demonstration, the transgenic and mutant fish lines used were:
- Tg(nyx:Gal4-VP16)q16a/+;Tg(UAS:gap43-YFP)q16b/+;Tg(UAS-E1b:NfsB-mCherry)c264/+; Tg(pax6-DF4:gap43-CFP)q01/+; roya9/a9
- Tg(nyx:Gal4-VP16)q16a/+;Tg(UAS:gap43-YFP)q16b/+;Tg(UAS-E1b:NfsB-mCherry)c264/+;Tg(gfap:GFP)mi2001/+;roya9/+
- Note: In fish “1”, a subpopulation of nyx-promoter defined retinal bipolar cells express a membrane-tagged YFP and/or a NTR-mCherry fusion reporter (most express both), and nearly all retinal cells express a membrane-tagged CFP. Fish “2” are as above except Müller glia cells express cytosolic GFP and global retinal CFP expression has been eliminated. Expression of the NTR-mCherry fusion renders nyx-defined bipolar cells susceptible to prodrug-induced ablation 3,5.
2. Selecting and Preparing Embryos/larvae for Live Imaging
- Mate transgenic/mutant lines of interest, collect eggs and incubate/maintain at 28.5°C in 0.3X Danieau’s solution (or ’embryo medium’ of choice).
- If not in an “albino” background, PTU should be added to inhibit tyrosinase activity prior to any evidence of pigmentation in the tissue of interest (e.g., ~16 hpf for the eye, 200 μM final). Note that treatment with PTU at concentrations exceeding 75 μM may cause toxicity and/or teratogenic effects. However, because the eye is highly pigmented, treatments below 200 μM are not adequate for high-resolution confocal imaging; under these conditions it is important to select healthy looking fish for imaging. For imaging other tissues, 75 μM PTU may be preferable. Also, as noted above, use of “albino” mutant lines obviates the need to treat embryos with PTU and is a preferred strategy for imaging at late larval stages.
- Once fluorescent reporters are evident, sort fish according to transgene expression and general health using a fluorescence stereo zoom microscope or similar.
- Prior to the first day of imaging, dissolve low melt agarose (LMA) in embryo medium to a final concentration of 0.5%, heat and mix to get into solution, store at 40°C.
- On the first day of imaging, prepare mounting solution: Add tricaine (an anesthetic, 756 μM final) and PTU (if necessary) to 0.5% LMA in embryo medium, mix gently, and return to 40°C.
- Anesthetize fish by transferring into embryo medium containing PTU and/or tricaine, allow time for fish to become non-responsive to touch (~3 min).
- Transfer individual fish into mounting solution, taking care not to dilute the agarose so that it can be reused. We use a micropipette with trimmed tip for transferring; this also helps to maintain a controlled volume (~30 μL). After acquiring, invert pipette and let fish settle to the bottom so that touching the tip to the mounting solution allows gravity transfer without need to dispel any liquid.
- After transfer, expel anesthetic solution from micropipette and use it to transfer the fish in a ~30 μL droplet of mounting solution to a Petri dish.
Note: This mounting method is only relevant for upright microscopy where long-working distance water immersion objectives will be used. For methods relevant to inverted microscopes, where coverslips are required, see Andersen et al., 2010; O’Brien et al., 2009; Graeden and Sive, 2009. - Return mounting solution to 40°C heating block.
- Using an eyelash brush or similar gently rotate fish into the desired orientation and position the region of interest in the center of the agarose droplet.
- Repeat steps 6 to 10 to mount the desired number of embryos per dish. For serial time-lapse experiments (see below) we typically mount six fish at a time and try to limit imaging sessions to 1 hour per group.
- While mounting other fish check back to make sure previous subjects maintain desired orientation until agarose solidifies (~3 min).
- After agarose has solidified, transport fish to confocal microscope stage and gently add embryo medium containing PTU and/or tricaine to the dish until all fish are completely submerged.
3. “Multicolor” in vivo Confocal Imaging
Note: We have tried to generalize the protocol below such that it provides relevant information for other microscope configurations. Specific references to Olympus and ImageJ software applications are in quotes. Our imaging system is an Olympus FV1000 upright confocal microscope equipped with 405, 440, 488, 515, 559, and 635 nm lasers, two variable barrier filter detection channels (allowing emission wavelength settings to be adjusted in 1 nm increments), one standard barrier filter channel (for red and far red imaging), and one transmitted light channel.
- Open the imaging software (“FV10-ASW”) and use either transmitted or fluorescent light sources to locate the region of interest. For optimal imaging of cellular and/or molecular detail use long-working distance objectives with high N.A. (e.g., 60X, 1.2NA).
- If collecting transmitted light images, focus the field diaphragm for Kohler illumination.
- Choose appropriate fluorphores from the “Dye List” or load acquisition parameters from a previously saved image file. Make adjustments to emission ranges (“Spectral Setting”) necessary for clean separation of reporter signals and/or maximal signal to noise ratios. These settings will need to be empirically defined for each experiment to minimize crosstalk but maximize signal; settings for the examples provided here are shown below:
Flourphores Lasers (nm) Barrier Settings / Emission Filter (nm) 1) ECFP, EYFP, mCherry* 440, 515, 559* 460-500, 530-545, 575-675 2) EGFP**, EYFP**, mCherry 488, 515, 559 500-515, 530-545, 575-675
*Imaging of mCherry could be improved using a higher wavelength laser (e.g., 568 or 594 nm)
**A sequential imaging mode allowing dichroic mirror and barrier filter changes (“Virtual Channels”) is required due to emission/excitation overlap between GFP and YFP. GFP and mCherry can be assigned to one channel, YFP to a separate channel. Note: crosstalk between GFP and YFP images will be evident prior to Image Subtraction (Part 6). - Make sure the objective being used is selected in the drop down menu to ensure any software defined presets are adjusted properly.
- To focus lasers and set power levels, select a look up table (LUT) that reveals pixel saturation (“Hi-lo”) for each channel. Set detector sensitivity (“HV”, PMT voltage) to a value consistent with desired image quality (defined empirically) and gradually raise laser output until image intensity is acceptable avoid pixel saturation.
- Check for crosstalk between channels; ensure that each laser line produces detectable signal only in the appropriate channel by manually turning lasers on and off while monitoring all imaging channels. To eliminate crosstalk reduce laser power. If no crosstalk is evident, all channels can be acquired simultaneously providing significant time savings.
- In the event of unavoidable crosstalk, use a “Sequential” imaging mode that switches between lasers/channels individually. Extreme cases (e.g., imaging GFP and YFP) require the use of sequential modes that also switch dichroic mirrors and/or barrier filters (“Virtual Channels”). Note: this option introduces significant temporal delays into the image acquisition process and may not be appropriate for capturing highly dynamic processes.
- “Zoom” and “Rotation” functions can be used to further frame the area of interest. Zoom can also be used to improve image resolution, up to the limits of the objective and scan format used (e.g., 512 x 512; for information about these issues see, http://corefacilities.systemsbiology.net/imaging/documents/app_note_image_res.pdf).
- For in vivo imaging in general, and of particular importance for time-lapse imaging, techniques which minimize phototoxicity issues (i.e., accumulation of free radicals) need to be emphasized; fast scan speeds (e.g., 2 μs/pixel) and low laser output levels (e.g., 1%) should be used whenever possible, as well as the lowest resolution scan format adequate for capturing salient information.
- To improve image resolution, scan averaging (“Kalman”) can be used during acquisition. Slower scan speeds also improve resolution but increase laser dwell time, which will exacerbate phototoxicity and bleaching issues. Note that both of these approaches are not ideal for time-lapse due to potential for inability to capture rapid changes across a z-series, a phenomenon we call “time blur”. Note that averaging by line versus by frame can help to eliminate “time blur” issues within a single plane but not across z-depths.
- If signal intensities are low and maximum image resolution is not required (e.g., simply counting cell numbers), increasing the confocal aperture (pinhole diameter) can be used to boost signal. This serves to keep laser intensities low, reducing phototoxicity and bleaching, however adjusting the aperture to a value greater than 1 airy unit leads to rapid losses in image quality.
- Define z-depth dimensions using a rapid scan mode (“Focus x4”). Because signal intensity generally decreases with z-depth it is best to start z-stack acquisitions at the lowest depth and end at the most superficial, this allows more difficult signals to be acquired before significant bleaching occurs.
- Check for adequate signal detection throughout the depth of the image. If desired, a z-dimension brightness correction function (“Bright Z”) can be used to adjust image acquisition parameters at specified depths.
- Set the z-dimension step size according to experimental needs. For instance, in our first example (Figure 1) we were simply taking a ‘cell census’ in individual retinas over successive days, thus we set the z-depth to 15 microns and took five to seven images per retina. This strategy allowed us to sample each retina with no cellular overlap between images. In our second example (Figure 2), we performed rapid 4D time-lapse movies in which GFP and mCherry signals needed to be spatially correlated, thus we set the step size to three times the theoretical lateral resolution a compromise which allowed z-stack images to be taken every 5-10 minutes but still served to resolve individual cellular detail.
- Acquire images (“XYLZt”) and save. Move on to the next fish if performing a serial time-lapse experiment (see #4 below). For rapid time-lapse imaging see #5 below.
4. Serial ‘Catch and Release’ Imaging: a Method for Tracking Cellular Changes in Individual Fish Over Successive Days
- A ‘catch and release’ protocol is used to minimize the amount of time fish are immobilized and to increase the number of fish imaged per experiment. The usefulness of this technique is limited to biological processes which unfold over days (e.g., Mumm et al. 20062). One advantage of this approach is that it allows any observed changes to be internally controlled; i.e., comparisons between different states of individuals rather than across populations. For instance, tracking changes in the number of labeled cells present in a given tissue can be performed accurately even if the number of cells initially labeled varies greatly between individual fish. Methods for using this protocol within the context of a prodrug-induced cell ablation assay 3,5 are provided here.
- To visualize the ablation and regeneration of targeted cell types in individual fish over time, confocal images are acquired prior to prodrug administration (Pre-treatment), immediately following prodrug removal (Post-treatment) and during the recovery period (Recovery images).
- Pre-treatment imaging: Fish are mounted and imaged as above. Once fish have been imaged they are released into embryo medium (+ prodrug) and incubated at 28.5°C until the Post-treatment imaging session. Working in teams of two, one mounting and releasing fish, the other imaging is a good way to maximize the number of fish that can be imaged per day.
- After imaging all fish in a single dish replace the embryo medium containing tricaine with embryo medium alone (+/- PTU).
- To release fish, use jewelers forceps to cut through agarose on both sides of the larvae and then gently tease agarose away until fish floats freely in embryo medium.
- Prodrug-induced ablation: Transfer larvae into an individual well of a multiwell plate containing embryo medium (+/- PTU) + prodrug (e.g. 10mM metronidazole, MTZ). To ensure accurate final prodrug concentrations we typically aliquot a defined volume of embryo medium (+/- PTU) containing a 2X concentration of prodrug (e.g., 20 mM) into each well. Fish are then added to wells in an equal volume of embryo medium (+/- PTU) to bring the final prodrug concentration to 1X.
- Incubate at 28.5°C until targeted cell ablation can be verified.
Note: Prodrug treatment regimens required to achieve successful ablation vary according to the cell type targeted; prodrug concentration and treatment duration will need to be determined empirically, be aware that high concentrations cause general toxicity, e.g., >10 mM MTZ. In addition, perdurance of fragmented reporter signals can complicate assessments of ablation success, in some instances confocal microscopy is required to adequately visualize cell loss. - Post-treatment imaging: Fish are mounted such that the same tissue is optimally oriented and imaged as above. Once all fish on a given plate have been imaged they are released into embryo medium (+/- PTU) into individual wells (as above) and incubated at 28.5°C to allow regeneration of the ablated cell type.
- Depending on the age of the fish and time required for recovery it may be necessary to feed fish during this stage of the assay. We use live food such as paramecia or rotifers to ensure optimal survival. It is also advisable to provide fresh media daily, particularly if fish are maintained in low volumes (e.g., 96 well plates).
- Recovery imaging: To document the kinetics of cell replacement, fish can be imaged on a daily basis during the recovery period. Once the kinetics have been established the assay can be simplified by acquiring recovery images only at time points of interest (e.g., full recovery). Figure 1 provides an example of this approach from our studies of retinal cell regeneration.
- At the conclusion of the experiment fish should be euthanized by immersion in 20X tricaine (15.3 μM) solution on ice.
5. Rapid Time-lapse Imaging.
- For rapid time-lapse imaging (i.e., ‘movies’) of multiple fluorphores, techniques which minimize phototoxicity issues are critical (see above). As stated above, this technique is best suited to fluorphores allowing simultaneous, rather than sequential, imaging
- Maintain fish at 28.5°C using a heated stage, ITO glass heating element, or similar.
- Use methods which reduce evaporation to help stabilize temperature and osmolarity (e.g., o-ring sealed coverslips for inverted microscopes).
- If using automated acquisition over extended times be sure to set the stack size to allow for growth and/or drift in the z-dimension.
- Set time intervals and number of scans (“TimeScan” or “TimeController”). Acquisition rates will need to be established empirically according to the dynamics of the biological process imaged and/or adjusted according the phototoxic sensitivity properties of the cell population of interest.
- Acquire images (“XYLZt” button) and save.
- To construct ‘movies’, areas of interest will need to be defined and aligned in the x, y, and z dimensions. The full process is beyond the scope of this protocol review but several software programs are available which also facilitate zoom, rotation, and panning functions during 4D image assembly (Volocity, Amira, etc.).
6. Spectral Separation Image Processing using Imagej Freeware
- Optimally, when performing a multi-reporter imaging experiment it is best to use fluorphores that are well separated spectrally. However, this is not always practical and/or possible. Here we provide a simplified method for separating fluorphores that have overlapping excitation and emission profiles, in this case GFP and YFP. In the example provided, we have used the time-lapse imaging techniques described above to track the behavior of GFP-labeled retinal ‘stem cells’, Müller glia, during the targeted ablation and regeneration of retinal bipolar cells labeled with YFP and NTR-mCherry (Figure 2).
- Collect images using sequential imaging modes and variable barrier filters that allow overlapping fluorophores to be acquired independently and partially separated, respectively (see above for GFP/YFP settings example). To remove crosstalk we use a simple image subtraction strategy which removes one channels signal from another.
- Image Subtraction Process: Install the latest version of ImageJ (http://rsb.info.nih.gov/ij/notes.html), Loci Bio-Formats tool (http://www.loci.wisc.edu/bio-formats/imagej), and align stacks plug-in (http://www.med.harvard.edu/JPNM/ij/plugins/Align3TP.html).
- Use the Loci Bio-Formats Import plug-in to open image files; simply dragging the file of interest onto the ImageJ window will start the import tool. Split the channels into individual stacks using the “Split Channels” checkbox in the import window or the “Split Channels” tab under “Channel Tools” (shift + ctrl + z) after loading.
- Start the Align 3 TP plugin (plugins>Align stacks> Align 3 TP). The stacks need to be aligned to ensure proper subtraction. Select the reference stack in the first dropdown box and the mis-aligned stack in the second. Check the “use xy relative origin” and “use z relative origin” boxes and click “OK”.
- To start the alignment process, click the “Volume Registration” button. A new window will appear that contains the alignment parameters. This section has been optimized by the distributor of the plugin, no modifications are necessary, simply click “OK”.
- When finished, an “Aligned Stacks” window will appear. Select “Output” from the Align window and the Output window should appear. This window is also optimized and needs no modification. Click “OK”. A new stack should appear in the work space with “Aligned” appended to the end of the file title.
- Crosstalk will be removed using the “Image Calculator” (Process>image Calculator). Select the stack to be subtracted in the Image1 dropdown box and the stack used in the subtraction in the Image2 dropdown box. Select “Subtract” in the Operation dropdown box and click “OK”. ImageJ will ask to process the stack, select “Yes”. A new stack should appear which has removed the crosstalk between overlapping channels.
- To recreate the same image structure before alignment and subtraction Stacks need to be merged. Select “Merge Channels” in the Channel Tools window. The tool will allow you to merge up to four stacks into a hyperstack.
Note: To merge more than 4 stacks, concatenate stacks in the desired order using the concatenate tool (images>stacks>tools>concatenate). Convert the concatenated stack to a hyperstack (images>hyperstack>stack to hyperstack). Concatenation of the stacks changes the depth, time, channel (zct) order, this needs to be reset within the dropdown box that appears; set “order” to xyztc, set “channels”, “slices” and “frames” according to the number of stacks, depth of the stack, and number of time points, respectively.. - Finally, all channels need to be colorized, adjusted for brightness and contrast, and saved as TIFF files. From here, conversion to RGB, montage, z-projection, or time series is the same process as for the original files.
7. Representative Results
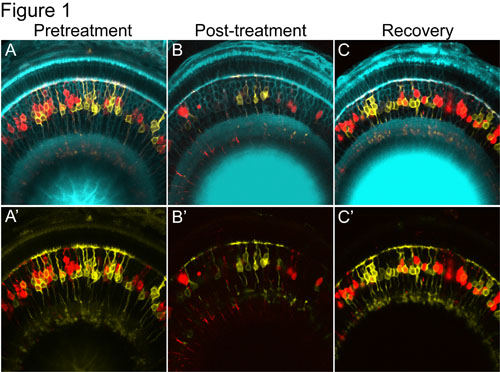
Figure 1. A time-lapse series of confocal images demonstrating targeted loss and regeneration of NTR-mCherry expressing retinal bipolar cells (red cells). A & A’) Prior to treatment there is mosaic expression of membrane-tagged YFP reporter in “control” bipolar cells, shown in yellow, and nitroreductase-Cherry fusion protein in “targeted” bipolars shown in red. B & B’) Following treatment with metronidazole, the red nitroreductase-expressing bipolar cells, are lost, while the yellow “control” bipolar cells, are spared. This demonstrates the highly specific nature of this ablation methodology. C & C’) When metronidazole is removed, NTR-expressing (red) cells return.
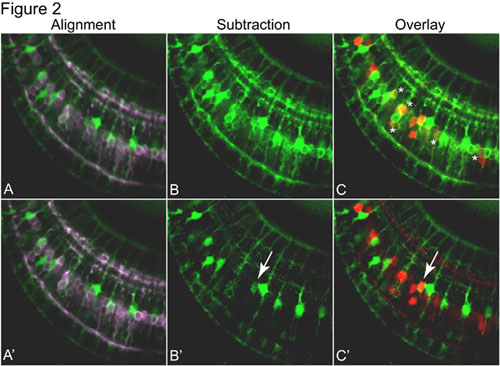
Figure 2. Provides an example of the image subtraction process that allows GFP and YFP to be cleanly separated following acquisition. A & A’) Prior to subtraction, the GFP (green) and YFP (pseudocolored purple) images are aligned. B & B’) The subtraction process allows YFP-labeled bipolar cells to be removed from the GFP image, allowing GFP-labeled Müller glia to be more easily detected. C & C’) Co-expression of GFP (green) and mCherry (red) is evident in the cell indicated by the arrow. Because Co-expression of the Müller glia cell marker and bipolar cell marker suggests that bipolar cell ablation triggers Müller glia to replace the lost cells. This interpretation is in keeping with recent studies of retinal regeneration which implicate Müller glia as injury-induced ‘stem cells’ in both mammals and fish.
