An In Vitro Technique to Stimulate Lymphocytes via Pathogenic Bacteria
Abstract
Source: Dousha, L., et al. Assessing Respiratory Immune Responses to Haemophilus Influenzae. J. Vis. Exp. (2021)
This video demonstrates an in vitro technique to stimulate lymphocytes via non-typeable Haemophilus influenzae (NTHi) and to assess the immune response. Incubating isolated peripheral blood mononuclear cells (PBMCs) with NTHi causes the bacterial cells to stimulate T lymphocytes, leading to cytokine production. Upon inhibiting the secretion of cytokines, immunofluorescence staining is performed to detect the accumulated intracellular cytokines in stimulated T lymphocytes.
Protocol
1. Antigenic preparation
NOTE: Three different antigenic preparations can be used to assess the immune response to Hi. These are 1) a subcellular component (typically from the bacterial cell wall); 2) killed and inactivated bacteria; and 3) live bacteria. Determine the use of each antigenic preparation prior to the initiation of any experiments.
- Subcellular components
- Obtain subcellular components from sources, including commercial preparations, in-house developed components, and/or from other investigators.
NOTE: Subcellular components usually from the bacterial cell wall may be used and include outer-membrane proteins such as P6 and lipooligosaccharide (LOS). These subcellular components are usually derived in specialized centers. The description of how they are made is beyond the scope of this article. Direct contact with experts in this field is recommended to obtain these components.
- Obtain subcellular components from sources, including commercial preparations, in-house developed components, and/or from other investigators.
- Killed/inactivated bacteria
- Obtain the bacteria from the appropriate sample (e.g., sputum or bronchoscopy). Confirm the strain as H. influenzae using an appropriate microbiology laboratory. Perform typing of the H. influenzae samples to confirm they are nontypeable (NTHi, by a specialist microbiology laboratory).
- To obtain a representative antigen, use multiple NTHi strains (at least 5, each of approximately the same amount) to make a pooled antigen. Store the strains at -70 °C in glycerol broth in microcentrifuge tubes.
NOTE: In this experiment, ten distinct strains were used. - Thaw the strains (one microcentrifuge tube at a time) out onto chocolate agar plates and grow overnight in a 37 °C incubator with 5% CO2. Add the bacteria to 500 μL of phosphate-buffered saline (PBS) and wash them twice (spin at 300 x g for 5 min). Use a MacFarland standard or a spectrophotometer to aliquot NTHi to a concentration of 108 mL (using a 5 mL container or equivalent).
- Heat inactivate the bacteria by placing them in a water bath at a temperature of 56 °C for 10 min.
- Sonicate the bacteria using a continuous sonication setting at a low-to-mid range speed for 30 seconds.
- Aliquot the appropriate volume of samples into microcentrifuge tubes. For a sample of 108 mL, use aliquots of 50 μL (i.e., 20 samples per mL), and freeze them at -70 °C until required.
- Live bacteria
- First, characterize live bacteria as mentioned in step 1.2.1. Use one well-characterized strain. Ensure that the strain is also stored in glycerol broth at -70 °C as a reserve.
- Grow the bacteria on enriched media such as chocolate agar plates or in broth.
- To use chocolate agar plates, spread the bacteria for a minimum of every 3-4 days with sterile spreaders.
- Alternatively, use Brain Heart Infusion (BHI) broth enriched with factors X (hemin) and V (β-nicotinamide adenine dinucleotide) to grow the bacteria. Inoculate NTHi from overnight plates in 5-10 mL of BHI broth supplemented with both hemin and β-nicotinamide adenine dinucleotide (both 10 µg/mL) and culture overnight at 37 °C in a 5% CO2 incubator.
NOTE: This has the advantage of reproducibility, higher colony-forming units (CFU) counts, and all bacteria being at a similar phase of log growth (on plates, bacterial viability can be greater depending on the position in the culture).
- Use a multiplicity of infection (MOI) of 100 bacteria in one cell to elicit a strong immune response while maintaining cellular viability. Use MacFarland Standard or spectrophotometer to assess the number of bacteria.
- Ensure that the media used for live NTHi assays is free of any human serum, as this will kill the bacteria. Animal serum samples (e.g., fetal calf serum) do not generally cause any problems.
NOTE: Use the methods described below to analyze standard tissue samples such as peripheral blood, bronchoscopy samples (particularly bronchoalveolar lavage (BAL)), and resected lung tissue. Incubate the samples with Hi antigen from 10 min to 24 h or more.
2. Assessment of lymphocyte function in peripheral blood
- Use whole blood or mononuclear cell preparations for flow cytometry assays.
- Acquire samples from each subject by venipuncture. Collect 4 mL of blood from a peripheral vein in a lithium heparin tube and divide the samples into aliquots for control and antigen stimulation.
- Add costimulatory antibodies (anti-CD28 and CD49d, 1 µL/mL) to both samples. Add NTHi to the antigen sample and incubate at 37 °C and 5% CO2 for 1 h. For the killed NTHi preparation, add 200 μL of the antigen to 2 mL of blood. For live NTHi, add live NTHi cells at an MOI of 100:1 to the white cells (as measured by hemocytometer).
- Add the Golgi-blocking agent Brefeldin A (10 µL/mL) to the samples and incubate them for another 5 h.
NOTE: Blocking the Golgi apparatus prevents the cytokines from being exported outside the cell. - If whole blood is used, lyse the erythrocytes with 0.8% ammonium chloride to leave only leukocytes in solution. Fix the leukocytes using 500 μL of 1%-2% paraformaldehyde for 1 h.
- Count the cells by hemocytometer, permeabilize 106 cells with 100 μL of 0.1% saponin for 15 min, and incubate the cells with fluorescent-labeled antibodies. Wash the cells and analyze them using a flow cytometer.
NOTE: The quantity of fluorescent-labeled antibodies will be specific for each cytokine. Follow the manufacturer's instructions. Typically, 106 cells in 100 μL of solution will be stained for 1 h. Most commercially obtained antibodies will be enough to perform 25-100 tests. - Determine the proportion of antigen-responding cells by gating the relevant lymphocyte population (cells are gated on by CD45, then by CD3, and later by CD4 or CD8 expression) as shown in Figure 1. Perform background staining on non-stimulated cells for all the cytokines to be analyzed. Screen 100,000 cells to analyze each cytokine for both stimulated cells and control.
Representative Results
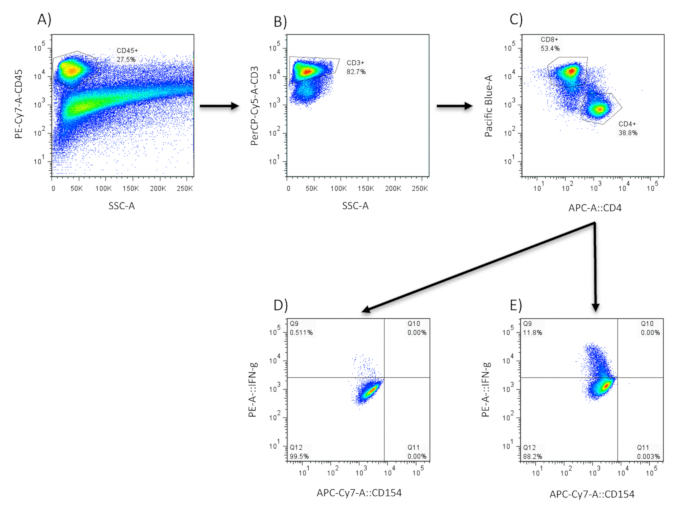
Figure 1: Cytokine production in lung tissue. Cells are first analyzed for their expression of the leukocyte marker CD45 (A) using flow cytometry. This population is then analyzed further for CD3 expression (B) and CD4/CD8 expression (C). CD3/CD4+ cells are assessed for intracellular cytokine production in control (D) and NTHi-stimulated samples (E).
Declarações
The authors have nothing to disclose.
Materials
| Ammonium chloride | Sigma Aldrich | 213330 | |
| Brefeldin | Sigma Aldrich | B6542 | |
| CD28 | Thermofisher | 16-0289-81 | |
| CD49d | Thermofisher | 534048 | |
| Saponin | Sigma Aldrich | 8047152 |
