1. Transfection of Cell Lines
Transfection Method 1
(Lonza Nucleofection for increased primary cell transfection efficiency)
- Cells should be of recent passage and in log phase growth prior to transfection. Wash cells in phophate buffered saline (PBS) and harvest cells by trypsinization.
- Collect cells by centrifugation at 1500 x g for 5 minutes.
- Add sterilized cover slips to a 6-well plate. Cover slips may be coated for enhanced cell adhesion. Place 1.5 mL of culture media (in this case Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS)) into each well and equilibrate the media in the incubator.
- Harvest the cells by trypsinization and resuspend in a minimal volume of phosphate buffered saline (PBS).
- Count the cells and centrifuge the correct amount of resuspension to provide ~5×10 5 cells per sample.
- Resuspend the cells carefully in 100 μL room temperature Nucleofector solution per sample.
- Combine 100 μL of cell suspension with 5 μg plasmid DNA and transfer the sample to a Nucleofector Cuvette being careful not to include air bubbles.
- Select the appropriate Nucleofector Program (this may require previous testing to establish optimal transfection efficiency with minimal mortality).
- Insert the cuvette containing the cell/DNA suspension into the Nucleofector Cuvette Holder and begin the selected program.
- Upon completion, remove the cuvette and add ~500 μL of pre-equilibrated culture media to it (taken from the 6-well plate).
- Immediately transfer the sample into the 6-well plate with final volume of 1.5 mL per well. Cells should be plated in either mixed populations or separately, for controls.
Transfection Method 2
(Qiagen Effectene transfection for cell lines)
- Prepare transfection complexes using the Qiagen Effectene Reagent by first adding 0.8 μg of each plasmid DNA sample to 200 μL EC buffer.
- Add 6.4 μL enhancer reagent to each sample, mix briefly by flicking the tube, and incubate for 2 minutes at room temperature.
- Add 20 μL Effectene reagent to each sample, mix by flicking the tube, and incubate for 15 minutes at room temperature.
- During this incubation, prepare the two cell lines of interest for transfection. Wash recently passaged cells (<80% confluence) once in phosphate buffered saline (PBS). Add back 1.5 mL fresh warmed and equilibrated growth medium (in this case Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS)).
- Once the transfection complexes have incubated for 15 minutes, transfer 1.2 mL of media to each sample. Mix by pipetting, and immediately transfer ~700 μL of the complexes to each of two wells in the 6-well plate. Add dropwise, and mix by gently by swirling the plate. Allow cells to transfect for at least 3 hours (may vary with cell type).
- Add sterilized cover slips to a new 6-well plate. Cover slips may be coated for enhanced cell adhesion.
- After the cells have transfected, wash cells twice with PBS and harvest the cells by minimal trysinization by adding approximately 100 μL trypsin solution to each well, briefly agitating the plate to fully coat each well, and immediately aspirating off the trypsin. Once the cells have lifted, add 1.5 mL fresh media.
- At this point, cells should be plated in either mixed or separate populations (for controls) onto the sterile cover slips.
- Allow cells to recover for 12 hours.
2. Cell Fusion
- Remove culture media from the cells and wash twice with PBS.
- At this point, it is recommended to treat the cells with cycloheximide (100 μg/mL) to inhibit protein synthesis. Allow the cells to incubate for 2 hours and then wash the cells twice with PBS.
- Fuse cells by exposing them to a solution of 50% polyethylene glycol 1000 (PEG 1000) in serum- free DMEM for 125 seconds at room temperature.
- Wash cells with PBS to remove the PEG 1000 solution, and add 2 mL freshly warmed and equilibrated media.
- Allow cells to incubate for 18 – 24 hours.
Fluorescence Microscopy
- Remove the culture media, and wash the cells twice in PBS.
- Fix cells by adding 1 mL of a 4% paraformaldehyde solution (in PBS) to each well. Gently agitate for 15 minutes.
- Wash the fixed cells twice in PBS.
- Carefully remove the cover slips from the plate, wick off excess PBS. Invert onto microscope slides loaded with one drop of mounting medium (90% glycerol, 100mM Tris pH 7.5, 2% DABCO (1,4-diazabicoyclo-octane) containing DAPI (4′,6-diamidino-2-phenylindole).
- Remove excess mounting medium from the slides, and seal cover slips with nail polish.
- Visualize the cells via confocal or epifluorescence microscopy.
3. Representative Results
Figure 2 shows an example heterokaryon experiment using primary fibroblasts and H1299 non-small cell lung carcinoma cells. The protein of interest in this experiment (Chicken Anemia Virus-VP3) displays primarily cytoplasmic localization in primary cell types and nuclear localization in transformed cell types as visualized by epifluorescence microscopy. The protein is expressed as a fusion to either GFP (pEGFP-N1 vector, Clontech) in primary foreskin fibroblasts (PFF) or dsRed (dsRed-N1 vector, Clontech) in H1299 cells. Control samples involving self-fusions (top two rows) display multinucleated cells with no change in steady-state localization patterns. Such changes could otherwise occur due to sensitivity to fusion conditions or the formation of syncitia. Heterokaryon fusions (bottom row) demonstrate nuclear entry of the GFP-fused primary cell-derived protein and colocalization with the dsRed-fused protein in response to the introduction of transformed cell material. The example shown is a relatively rare heterokaryon fusion resulting from only two cells, one of each type, as demonstrated by the presence of both fluoroprotein signals. In addition to detecting these types of localization transitions, nucleocytoplasmic shuttling activity can be easily detected by the presence of a single fluorophore in both nuclei. This type of determination is clear when examining 1:1 cell fusions and would depend on inhibition of translation.
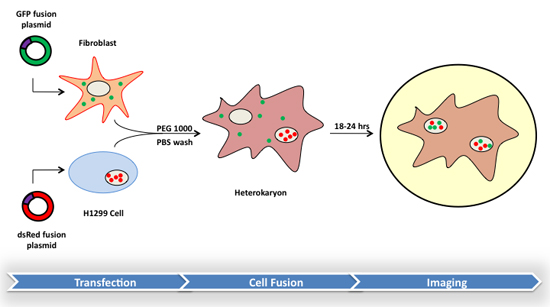
Figure 1. Flow chart of the heterokaryon method using primary fibroblasts and H1299 non-small cell carcinoma cells. The gene of interest is cloned to produce an in-frame fusion to one of two fluorescent protein genes. Cell lines are then transiently transfected with either construct and allowed to recover. Heterokaryon formation is induced by brief treatment with polyethylene glycol (PEG) followed by further incubation. Finally, the sub-cellular localization of the protein of interest is observed using various forms of fluorescent microscopy.
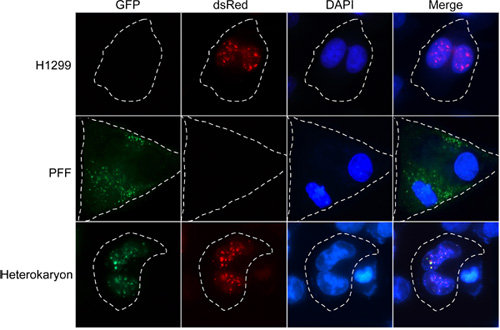
Figure 2. Nucleocytoplasmic trafficking and shuttling activity of the Chicken Anemia Virus VP3 protein visualized by heterokaryon fusion. Top row: Representative images of self-fused H1299 cells expressing dsRed-VP3. Middle row: Representative epifluorescence images of primary foreskin fibroblast cells (PFF) expressing GFP-VP3 after PEG fusion. Bottom row: Heterokaryon fusion of PFF and H1299 cells. Yellow indicates colocalization of GFP and dsRed signals. Cells were imaged using a Leica AF6000E fluorescence microscope and Leica imaging software.
