1. Sample Preparation
- Plant materials are harvested and frozen immediately.
- Frozen plant materials are powdered by mixer mill (or mortar) and stored in Falcon tube or Eppendorf tube at -80 °C.
2. Extraction for Metabolite Profiling
- Aliquot frozen plant material in a 2 ml Eppendorf tube.
- Add 5 μl of extraction buffer per milligram of fresh weight of frozen plant material.
- Add one metal (or gilconia) ball and homogenize of frozen powder with the Mixer Mill for 2 min at 25 l s-1.
- Centrifuge 10 min at 12,000 rpm.
- Transfer the supernatant to NANOSEP centrifugal filter.
- Centrifuge 2 min at 4,000 rpm.
- Transfer the supernatant to new Eppendorf tube, and store at -20 °C until use.
3. Metabolite Profiling by LC-MS
- Set up HPLC and check temperature of column oven and sample tray.
- Set up MS condition and check the state of vacuum and heating capillary.
- Perform m/z calibration of MS detector.
- Transfer of 50 μl of extracts to glass vial for LC-MS.
- Inject 5 μl of extracts to LC-MS.
4. Data Analysis
- Configure Xcalibur or Metalign 4 and select the data analysis to be processed.
- Prepare a table of detected peaks of your interest in accordance with compound class in Table I.
- Identify peaks by co-elution of standard compounds.
- Annotate detected peaks using MS2 analysis, literature survey, metabolite database search (Figure 2, 12,13).
5. Prediction of Metabolic Pathway
- Construct a possible pathway with detected compounds. Prediction of pathway using peak annotations should be based on the chemical structure of detected compounds by predicting linking enzymatic functions on the metabolic pathway 13. Structuring biosynthetic steps should be conducted with precise peak annotation. But determination of detailed chemical structure, for example sugar moiety, is not indispensable in this step, because prediction of sugar moiety and adducted position is very difficult to identify by MS analysis. Determination of sugar type such as hexoside and pentoside will be identified by enzymatic assay of sugar donor at the last step of project. Mostly constructing of prediction of pathway should be performed as small molecule is intermediate of larger molecule except in the some cases such as dehydration reaction. List of atomic molecular weight, for example 16 m/z for difference between -H and -OH moiety (oxidation), 14 m/z (Carbon atom) for difference between -OH and -OMe (methylation) and 162 m/z (MW-H2O) for hexose (glycosylation), is useful for prediction. Determination of modification type with correlation analysis of tissues specificities helps prediction of metabolic pathway. Database of general metabolic pathway such as KEGG database (http://www.genome.jp/kegg/) and PlantCyc (http://plantcyc.org/), is very effective for prediction of metabolic pathway of your interest.
6. Preparation of Gene List with Arabidopsis Orthologous Gene ID
- Download gene ID list from genomic database.
- Add Arabidopsis gene ID of orthologous gene, in case of your target plant is not Arabidopsis.
- Prepare a list of genes in your pathway-of-interest. Annotation of Arabidopsis pathway data and gene family data are available in TAIR website (http://www.arabidopsis.org/). If you prepared a list of Arabidopsis orthologous genes, you can subsequently combine them.
7. Co-expressed Gene Analysis
- Test using the prepared gene ID list to search best database for your pathway by checking the correlation using well known gene pairs in your pathway-of-interest (Table II). If co-expression database or gene expression database are not available in the plant of your interest, Arabidopsis co-expression database should be used with a list of Arabidopsis orthologous genes. In case of barley, rice, poplar, wheat, medicago and soybean, co-expression analysis of plant species can be used (Table II).
- Construct the framework for your target co-expression network based on the connections of the well-known genes in your pathway-of-interest.
- Add correlated candidate genes (r<0.4~0.90, within approximate value of coefficient value between the connections of the well-known genes in your pathway-of-interest) and check their gene annotation in your predicted families to the connections of this network for finding best candidate genes (Figure 3). Threshold of coefficient value should be coordinated according to network structure and density of correlated genes.
- Make list of genes which you are able to narrow down as being specialized to your target pathway.
- Check gene expression of the organ specificities and stress responses of your candidate genes.
8. Integration of All Information to Predict New Pathways
- Add well-characterised genes which have been used for query of co-expression analysis to predicted metabolic pathway.
- Check uncharacterized parts in this pathway, for example uncharacterized enzymatic steps, transport proteins and transcription factors.
- Predict most suitable gene annotation for these missing uncharacterized steps.
- Combine the results of metabolite profiling and candidate genes of in silico gene expression based on the predicted pathway.
- Arrange your candidate genes on the predicted pathway according to gene function, for example, acetyltransferase for acetylated metabolite, glycosyltransferase for glycoside, P450 for oxidised compound. Phylogenetic tree analysis of amino acid sequences is useful for some wide gene family such as P450 and glycosyltransferase.
- Check the consistency of tissue specificities or stress responses between metabolite accumulation and gene expression level of candidate genes.
- Check the connections to other metabolism for providing substrate and stress responsive genes.
9. Experiments for Gene Identification Using Bio-resources
- Check the availability of bioresources for facilitation of experiment for candidate gene identification.
- Perform an experiment for identification of gene function using bio-resources, such as KO mutant library and full-length cDNA library. The experiments for functional identification of genes with preparation of overexpression plants and knockout mutants, enzymatic assay and promoter binding assay, have to be performed for the best candidate genes in your prediction. Recombinant protein assay for characterisation of protein properties and preparation of overexpression plants better to be carried out after confirmation of metabolite profile using KO mutant since it takes considerably longer to prepare recombinant protein and gene cloning for transformation.
10. Representative Results
The procedure of integrated analysis described in this protocol has many possibilities depending on specified experimental purpose and choice of biological and analytical combinations. Choice of procedures and experimental design should be carried out properly on the basis of your target pathway, compounds and plant species. The integration strategy described in this protocol is focused on annotation of plant gene function and the discovery of novel gene functions with an efficient usage of several bio- and data-resource. Expected outcome is promised to provide with the only case of conclusive prediction. This fact indicates that if enough evidences can not be given by combination profiles, experiment should not be started. For this reason, in any cases, additional preliminary experiments such as targeted gene expression profiling by RT-PCR, can support your prediction of gene function. Accuracy and correctness of prediction correlates higher depending on qualitative difference and number of variation of combination. In addition, good candidates and valid outcomes can only come from accurate prediction of pathways. Peak annotation should be conducted by combination of several approaches, for example literature survey, reference plant extract, MSn analysis, organ specificity and mutant analysis 13.
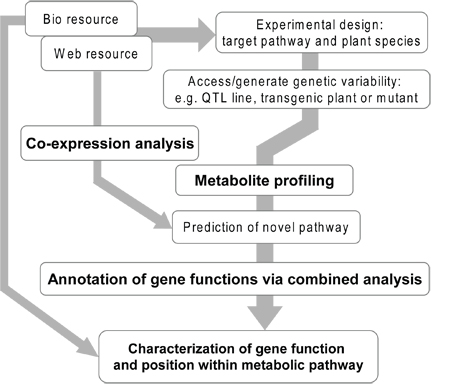
Figure 1. Overview of the experimental flow of gene annotation via combined approach. In some cases, projects start with the discovery of a novel peak which is detected in special conditions or tissues, and the desire to understand its role within its metabolism. In other instances the purpose of the project is gene identification or discovery of key regulatory factors such as transcription factors. Design of experiment should be planed with an data set which shows clear differences of metabolite levels in your target pathway, using a wide range of tissue samples from different organs, and for differentially grown plants or plants exposed to stress conditions, and subjecting the material to metabolite profiling. Mutant and transgenic plants as well as QTL harbouring breeding material also represent suitable genetic material for these studies. Prediction of novel pathway should be performed carefully with accurate peak annotation and combination approach with different type of metabolotype such as organ securities and stress responses according to gene expression data of your pathway-of-interest. In the last step, metabolite and transcript profiling should be performed which will eventually, when combined with in silico analysis of web-resources and in vitro characterisation of gene expression via heterologous expression, lead to the confirmation of the gene candidate and elucidation of its function and position within a metabolic pathway. Abbreviations: QTL, Quantitative Trait Loci.
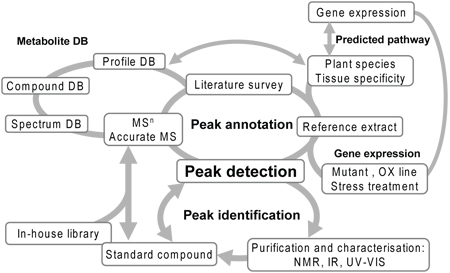
Figure 2. Work flow of combinational approach for peak annotation. An procedure for peak identification and annotation by the standard compound, comparison of wild type and knock out mutants, multi-dimensional mass spectrometry of the target peak referring to mass spectra of pure compounds from the databases 12. Abbreviations: DB, database; KO, knock-out; 1-D, one- dimensional; 2-D, two-dimensional; NMR, nuclear magnetic resonance; IR, infra-red; MSn, mass-mass spectrometries.
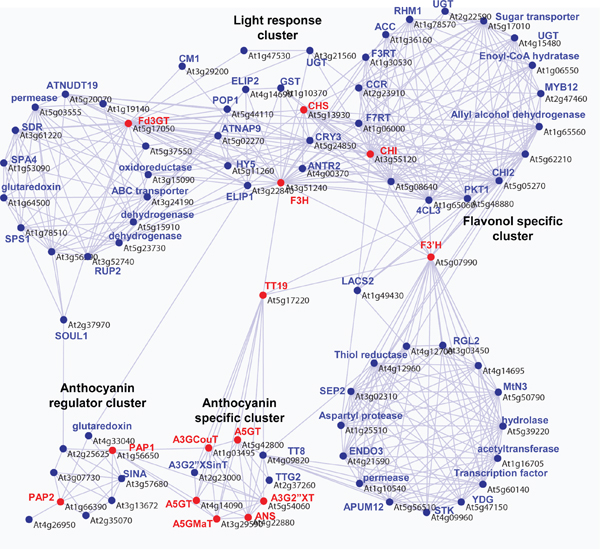
Figure 3. Example co-regulation network analysis of the anthocyanin pathway. Coexpression analyses were performed using the PRIMe (http://prime.psc.riken.jp/?action=coexpression_index) based on the data set of ATTEDII version 3 8,2 with the Pajek program (http://vlado.fmf.uni-lj.si/pub/networks/pajek/). Positive correlations (r<0.5) are used to make network connections. Red node: twelve anthocyanin enzymatic genes (At5g13930, CHS, TT4, chalcone synthase; At3g55120, CHI, TT5, chalcone isomerise; At3g51240, F3H, TT6, flavanone 3-hydroxylase; At5g07990, F3’H, TT7, flavonoid 3′-hydroxylase; At5g17050, Fd3GT, UGT78D2, flavonoid 3-O-glucosyltransferase; At5g17220, AtGSTF12, TT19; At5g42800, DFR, TT3, dihydroflavonol reductase; At4g22880, ANS/LDOX, TT18, anthocyanidin synthese; At4g14090, A5GT, anthocyanin 5-O-glucosyltransferase; At5g54060, A3G2″XT, putative anthocyanin 3-O-glucoside 2″-O-xylosyltransferase; At3g29590, A5GMaT, anthocyanin 5-O-glucoside 6”’-O-malonyltransferase; At1g03940, A3GCouT, anthocyanin 3-O-glucoside 6″-O–p-coumaroyltransferase) and two transcription factors for anthocyanin production (At1g56650, PAP1; At1g66390, PAP2) was used for searching candidate genes. Candidate genes were found by an “intersection of sets” search with a threshold value with a coefficient of r>0.50 queried by intersection of sets by all genes queried (Fourteen anthocyanin biosynthetic genes). A co-expression network, including correlated candidate genes (68 genes) and queried genes (14 genes), was re-constructed by an “interconnection of sets” search with r>0.50 using the PRIMe database. The output files that were formatted with a ‘.net’ file from the PRIMe database and networks were drawn using Pajek software. Blue node indicates candidate genes which correlated with anthocyanin genes.
| species | Major secondary metabolite |
| Arabidopsis thaliana | Glucosinolate, flavonol, anthocyanin, sinapoyl derivative |
| Populus trichocarpa | Flavonol, anthocyanin, salicylate derivative |
| Vitis vinifera | Flavonol, anthocyanin, tannin, stilbene |
| Solanum lycopersicum | Flavonol, anthocyanin, glycoalkaloid, chrologenate related, |
| Nicotiana tabacum | Flavonol, anthocyanin, nicotianamide, chrologenate related, acylsugar |
| Oryza sativa | Glycoflavone, anthocyanin, sterol derivatives |
| Zea may | Glycoflavone, anthocyanin, benzoxazinone, sterol derivatives |
| Medicago truncatula | Isoflavone, anthocyanin, saponin, |
| Lotus japonica | Isoflavone, flavonol, anthocyanin, saponin, |
Table I. Major secondary metabolites in model plant species.
| Co-expression database | Address |
| Plant cross species | |
| COP | http://webs2.kazusa.or.jp/kagiana/cop0911/ |
| PlaNet | http://aranet.mpimp-golm.mpg.de/ |
| Plant species | |
| ATEED-II | http://atted.jp/ |
| BAR | http://142.150.214.117/welcome.htm |
| COP | http://webs2.kazusa.or.jp/kagiana/cop |
| GeneCAT | http://genecat.mpg.de/ |
| Arabidopsis | |
| ACT | http://www.arabidopsis.leeds.ac.uk/act/coexpanalyser |
| AthCoR@CSB.DB | http://csbdb.mpimp-golm.mpg.de/csbdb/dbcor/ath.html |
| CressExpress | http://cressexpress.org/ |
| PRIMe | http://prime.psc.riken.jp/?action=coexpression_index |
| Oryza sativa | |
| RiceArrayNet | http://arraynet.mju.ac.kr/arraynet/ |
| Rice Array Database | http://www.ricearray.org/coexpression/coexpression.shtml |
Table II. Available gene expression database for in silico co-expression analysis.
