We carried out a set of transformations with E. coli and V. cholerae to compare transformation efficiency of our rapid methodology with an adaptation of the (traditional) method originally described by Dower et al.2 for the preparation of electrocompetent bacteria. To carry out the traditional preparation of competent cells, 500 ml LB in a 2 L-Erlenmeyer flask were inoculated with 500 μl overnight culture of E. coli DH5α or V. cholerae O395, incubated in an orbital shaker (215 rpm at 37 °C) and harvested at OD600 of between 0.5-1.0. The flask was cooled on ice and the cultures centrifuged in a pre-chilled rotor at 4,000 x g for 10 min at 4 °C. The cell pellets were thoroughly resuspended in 500 ml ice-cold 2 mM CaCl2 for V. cholerae and ddH2O for E. coli, and centrifuged again under the same conditions. Subsequent rounds of resuspension and centrifugation were carried out in decreasing volumes of 200 and 100 ml volumes making sure the culture remained chilled at 4 °C. Finally, a resuspension and centrifugation in a 10 ml volume was carried out with 10% glycerol. The final pellet was resuspended in 2 ml 10% glycerol and 40 μl aliquots were frozen at -80 °C and stored no longer than one week prior to electroporation.
Electroporation conditions including pulse settings and cuvette size were identical for both bacterial species in all electroporations regardless of method of preparation of competent cells. The only difference was that we carried out the washes in cold ddH2O for E. coli and cold 2 mM CaCl2 for V. cholerae. Table 1 shows results of bacteria transformed by electroporation identically but rendered competent either with the rapid protocol or with the traditional method. We employed strain O395 and DH5α as commonly used, representative strains of V. cholerae and E. coli respectively; similar results can be obtained with other strains of the same species (albeit not every single strain has been tested) and likely adapted to other Gram-negative bacteria and probably beyond.
To insure that equal cell numbers were present in each batch pulsed, electrocompetent bacteria generated utilizing the rapid method were pooled upon collection, mixed, kept homogenously suspended, and aliquoted in 40 μl volumes shortly prior to electroporation. Electrocompetent bacteria generated utilizing the traditional method were treated similarly prior to freezing in 40 μl aliquots. Following delivery of the pulse, each batch of 40 μl electroporated bacteria was suspended into 400 μl LB and serially diluted 1:10. One hundred microliters of each dilution were plated on LB agar in the presence of 100 μg/ml ampicillin and spread using a sterile glass cell spreader and incubated at 37 °C overnight. To quantify the total number of bacteria employed in each transformation, 40 μl volumes of electrocompetent bacteria of each prepared batch were serially diluted, and plated on LB agar without antibiotics identically in parallel. One aliquot of equally treated bacteria from each batch was electroporated with 1 μl Tris-EDTA (TE) buffer but without DNA (mock transformation), serially diluted and plated on LB agar without antibiotics to estimate the number of cells lost by the delivery of the electric pulse. Lastly, one additional variation to the experiment was carried out to determine whether 30 min incubations at 37 °C in 1 ml LB after electroporation but prior to plating on LB-agar would affect the recovery of transformed cells. colony forming units (CFUs) were enumerated on the following day.
For this experiment we employed pUC18, a common laboratory plasmid, and batches of competent bacteria consisting of approximately 108 CFUs for cells prepared utilizing the traditional method and 107 CFUs for cells prepared utilizing the rapid protocol. Electroporation alone (without DNA) reduced viable CFUs by 10-100 fold irrespective of the method used to prepare the electrocompetent cells. As shown in Table 1, transformant yield was within the 106 -107 CFUs/μg of DNA range in three replicate experiments (standard deviations in parentheses) for E. coli (DH5α) and V. cholerae (O395) respectively using the traditional, lengthier method for electrocompetent cell preparation. Transformant yield appeared decreased 10 fold to 105 -106 CFUs /μg of DNA in three replicate experiments (standard deviation in parentheses) for E. coli and V. cholerae respectively using the rapid method. For this reason we determined the percentage of transformed bacteria by dividing transformed CFUs by the number of CFUs recovered from “mock transformations” (without plasmid) from otherwise identical batches of competent cells. The percentage of transformed bacteria was within one log (2.5-9.4%) regardless of method of preparation and species transformed (Table 1 bracketed numbers). These results suggest that the efficiency of the rapid method is comparable to that of the traditional lengthier procedure of preparing competent cells and that the representative strains of Vibrio cholerae and Escherichia coli are equally amenable to the rapid procedure.
We found that transformation efficiency, for plasmids like pUC18 harboring β-lactamase cassettes, is not affected by the 30 min recovery time in LB broth. The same number of transformants was recovered whether the cells were plated to LB agar in the presence of ampicillin directly, after electroporation, or whether they were allowed a 30-45 min recovery time in LB broth at 37 °C prior to plating. Our findings that ampicillin-resistance does not require outgrowth under nonselective conditions suggests that β-lactamase expression occurs sufficiently fast upon transformation. However, it should be noted that we carried out serial dilutions of pulsed bacteria in LB broth, which may have contributed to recovery from the electric shock and initiate gene expression.
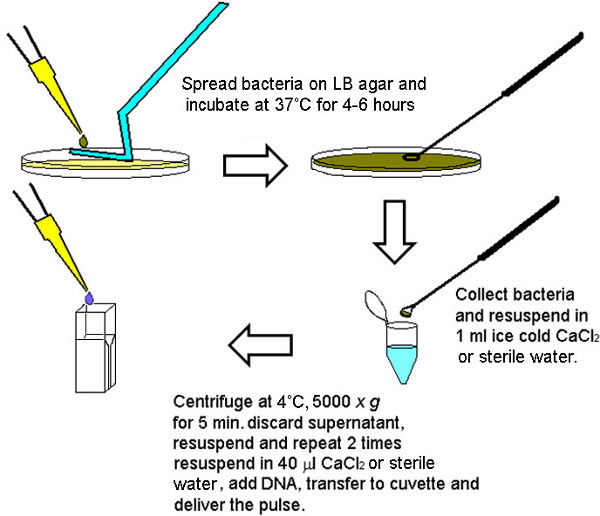
Figure 1. Graphical depiction of the methodology for the rapid preparation of electrocompetent bacteria. After 4-6 hr (depending on the density of the inocula) sufficient numbers of bacteria can be collected to carry out four to six transformations from a single LB agar plate. To insure equal number of competent cells for each final volume to be electroporated, bacteria collected from the LB agar plate were initially pooled and washed together then aliquoted in 40 μl volumes depending on the size of the pellet (larger pellets were diluted in larger volumes divisible by 40 μl).
| Number of Ampicillin-resistant transformants/μg DNA (Ampicillin-resistant transformants recovered [%]) | ||
| Strain | Traditional method | Rapid method |
| E. coli (DH5α) | 2.3 x 106 (± 7.3 x 105) [9.4%] | 3 x 106 (± 7.1 x 105) [2.5%] |
| V. cholerae (O395) | 4 x 107 (± 1.7 x 107) [5%] | 6.7 x 105 (± 2.1 x 105) [3.3%] |
Table 1. Transformation efficiency per microgram of DNA (pUC18) of electrocompetent bacteria prepared by the traditional method compared to the rapid method.
All transformations were carried out with 500 ng pUC18 DNA (~80% supercoiled DNA as visualized by 1% agarose gel electrophoresis) and the electroporated cells were serially diluted 10-fold in LB broth prior to plating for CFU on LB-agar with antibiotics. Controls of non-electroporated bacteria and of bacteria electroporated without plasmid, serially diluted and plated on LB agar without antibiotics were carried out for each set of transformations to determine the total number of viable bacteria prior and after pulsing. The electroporated controls represented the base line for the percent of viable bacteria successfully transformed.
