The approach described here was developed using specific instruments, tools and software. Because labs will not be using the same experimental setup the approach is generalized where possible.
1. Rapid Freezing
The analytical method to be described is absolutely dependent on cryogenic approaches for: 1) the "cryofixation" of cells or tissues in a manner that quantitatively preserves the distribution of diffusible tissue components and chemical elements as they were in live cells at the instant of freezing; and 2) the preparation of ultrathin cryosections suitable for imaging and analysis in a TEM. These techniques are briefly described here, but details necessary to reproduce these procedures in other laboratories are beyond the scope of this article. Interested readers are referred to excellent recent articles9,14.
Caution: This section describes the use of liquefied ethane, which is highly flammable and potentially explosive; appropriate precautions should be taken. Operator must also be familiar with liquid nitrogen (LN2) safety precautions, including protective lab coat, glasses, and cryoresistant gloves. Note: Here and throughout, all tools (forceps, etc.) used to handle frozen specimens must be precooled in LN2 before use to avoid accidental thawing.
Cultured cells on coverslips
- Prepare a plunge freezing device by condensing gaseous ethane at -160 °C in the LN2-cooled central well. Fill to the mark and cover the well with the pivoting aluminum lid when not in use.
- Using filter paper wedges, blot plastic coverslips of cultured cells under experimentally appropriate conditions to a thin aqueous film. Blot from the edge so as to avoid touching the tissue; the ideal residual film, determined by trial-and-error, is as thin as possible but not so thin as to evaporate and allow drying of the tissue surface.
- Holding the coverslip by the edge with clamping forceps, quickly immerse in liquid ethane, either manually or using the gravity-powered plunger.
- Place the frozen coverslip in a nearby styrofoam bowl of LN2, large enough to manipulate the desired number of coverslips into long-term storage containers. Aluminum screw cap cans that don't seize under LN2 are recommended.
Rapid freezing of cultured brain slices
- Under a dissecting microscope, view and orient an organotypic hippocampal slice, undisturbed and still attached to the culture membrane insert. Place fiducial marks on the membrane near the edge of the slice with a black Sharpie-type pen and photograph.
- Still under the microscope, cut out approx. 5 x 5 mm membrane squares with individual slices centered on the squares. Avoid touching the slice or bending the membrane.
- Mount a membrane-supported slice, cushioned by a ¼ in diameter agar pad, on a custom designed aluminum disk made to fit a cryoultramicrotome (described below). Rapidly freeze the slice by pneumatically propelling it against an LN2-cooled sapphire block by means of a customized "slam freezing" device.
- Move to storage as described for cultured cells.
2. Cryosectioning
CAVEAT: Successfully producing dry-cut ribbons of ultrathin sections, while straightforward and logical, requires training, patience and considerable practice.
- Bake out a cryoultramicrotome equipped with a cryoattachment and cool to -135 °C.
- Cut frozen coverslips into approx. 3 x 3 mm squares using a sharp, precooled scalpel. Embed pieces with cultures facing up in a viscous liquid "cryoglue" (a 1:6 mixture of ethanol and 2-propanol 11), on standard 3 mm diameter aluminum pins designed to fit the microtome specimen chuck. Avoid leaking cryoglue onto culture surface. Solidify cryoglue by lowering the cryobox temperature to ≤ -160 °C.
- For frozen brain slices, securely attach discs directly to a custom-made specimen chuck by means of a screw collar.
- Trim selected areas of the frozen specimens – e.g. cell-rich areas of coverslip pieces or identified areas of slices – to an approx. 250 x 250 μm block face and ~100 μm depth using a diamond trimming tool.
- Dry-cut ribbons of thin sections at ca. -160 °C using a 35° diamond cryoknife, essentially as described in referencs4,9. Cutting speed and knife clearance angle are determined empirically; a good starting point is 0.4-0.6 mm/sec at 9°. The nominal thickness, i.e. specimen advance, of hydrated sections is typically 80 nm, although sections are actually 1.5-2.0x thicker, mainly due to compression. For satisfactory sectioning, an antistatic device is essential. Place the ionizing tip of the device 0.5-1 cm from the knife edge and adjust output power until satisfactory ribbons of sections are produced.
- Prepare eyelash probes by gluing (with epoxy) eyelashes to wood applicator sticks. Using such a probe, pick up and transfer sections from the back of the knife onto glow-discharged, carbon-coated pioloform support films cast over 100-mesh folding copper grids and resting on a half-folded indium foil envelope on a working shelf behind the knife. Fold over the top half of the grid and envelope, press with a cold pressing tool.
- Transfer wrapped grids to a convenient grid box and store in an aluminum can as described in step 1.4.
3. Cryotransfer of Specimens to the Electron Microscope
The core instrument for EPMA in this laboratory is an analytical electron microscope operated at 120 kV and equipped for cryomicroscopy, that is, designed with a clean vacuum, specimen-area anticontaminator, a 2k x 2k high-sensitivity digital camera and cryotransfer specimen holder. Check beforehand microscope alignment and operating conditions in both low- and high-magnification modes and confirm a satisfactory column vacuum, ideally ≤10-7 Torr. Tune up as necessary.
- Confirm that the vacuum insulation of the Dewar component of the cryoholder is satisfactory. Pump as necessary.
- Cool the cryoholder to its minimum temperature, at least -160 °C, while under high vacuum within the microscope stage; detach the cable connecting the holder to its control box. Tilt the goniometer 45° clockwise before removing the holder to minimize LN2 spilling onto operator and microscope surfaces.
- Prepare the benchtop cryoworkstation by cooling the integral insulated cup to ≤160 °C.
- Transfer (quickly!) the cooled cryoholder from microscope to cryo workstation. Retract the holder's frost shield.
- Retrieve under LN2 a grid sandwich from storage and place on the working table of the cryoworkstation. A retaining ring for the holder's specimen well is also placed on the table.
- Open the indium envelope and move the enclosed folding grid to the specimen well of the cryoholder. Secure the grid with retaining ring using the spanner tool provided and close the frost shield.
- Quickly remove the cryoholder from the cryoworkstation and insert into the airlock of the microscope and go through the pumping sequence as quickly as possible. On insertion, there should be minimal disruption of the column vacuum.
- Return goniometer to 0° tilt. Reconnect and reenergize the control box and confirm that the specimen temperature is ≤150 °C.
- Refill the Dewar of the specimen holder and allow vacuum and temperature to fully recover.
4. Visual Survey of Sections
- Retract the frost shield of the holder to expose specimen and turn on the electron beam.
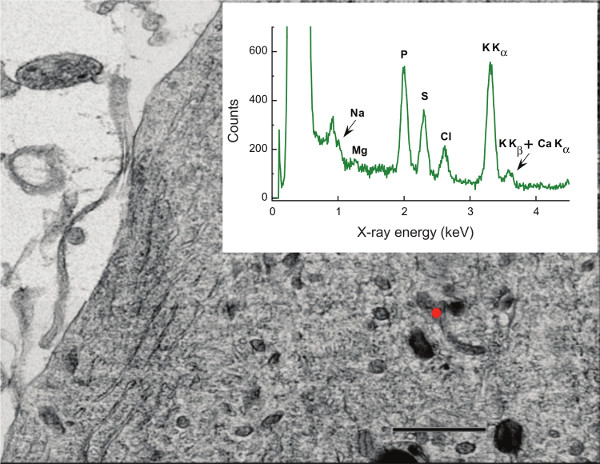
- Visually evaluate the specimen at low magnification, typically 250X, and low illumination. As illustrated in Figure 1, sections should be thin and smooth, not folded or overlapping, flat and well attached to the support film, and generally not obscured by grid bars.
- Optionally photograph selected sections. (NOTE: Minimize beam exposure, since frozen-hydrated sections are very susceptible to beam-induced damage.) Use the automated digital goniometer stage to store coordinates of selected sections.
5. Freeze-drying of Sections
- Freeze-dry sections by increasing holder temperature to ca. -100 °C for ~30 min.
- Recool holder to -160 °C or below.
- Before imaging, turn off cryoholder control unit and physically disconnect controller cable to avoid image drift due to thermal cycling and/or vibration pickup.
6. Imaging of Cells and Organelles
Structural images of freeze-dried sections are obtained at ca. -160 °C as low-dose, zero-loss images recorded digitally using a 2k x 2k slow-scan CCD camera controlled by appropriate software.
- Activate the EM in hi-mag mode.
- Choose and image at ~2,000X (as TIFFs, see Figure 2) selected cells and subcellular areas of interest in high-quality sections whose locations were previously recorded and stored. (NOTE: The dried sections are now substantially less fragile and less susceptible to electron beam damage.
- Evaluate images in order to select regions of interest (ROIs) for X-ray analysis. This step can optionally be performed off-line, in which case the EM can be turned off and the specimen allowed to warm to room temperature.
7. Acquisition of X-ray Spectra
X-ray spectra can be recorded using any of several commercial or custom-designed X-ray analysis systems minimally consisting of an energy-dispersive X-ray (EDX) detector, associated pulse-processor electronics and compatible acquisition and display software. (The system used in this lab is described in Table 1.)
- Configure the EM for the X-ray acquisition by inserting the EDX detector into the column (if necessary), withdrawing the objective aperture, and inserting and centering any stray radiation apertures. Adjust the cryoholder to the lowest temperature at which frost contamination of the specimen is avoided, but at least below -100 °C.
- Tilt the holder 20° toward the detector.
- Under wide-field imaging conditions, and assuming one intends to analyze the matrix of an individual mitochondria, choose a mitochondrion for analysis, move it to the center of the field and focus.
- Go to spot mode (on some microscopes just converge the beam using second condenser focus) and increase the beam current to ca. 3 nA (as measured with a Faraday cup or similar) into a 100 nm spot.
- Launch the spectrum acquisition software and begin 100 sec acquisitions, which can be viewed live time on the display monitor. Save recorded spectrum (Figure 2). The industry standard EMSA format is preferred.
- Shut down the EM and transfer the multiple-spectra file(s) to an (offline) analysis workstation.
8. Analysis of X-ray Spectra
Qualitative analysis
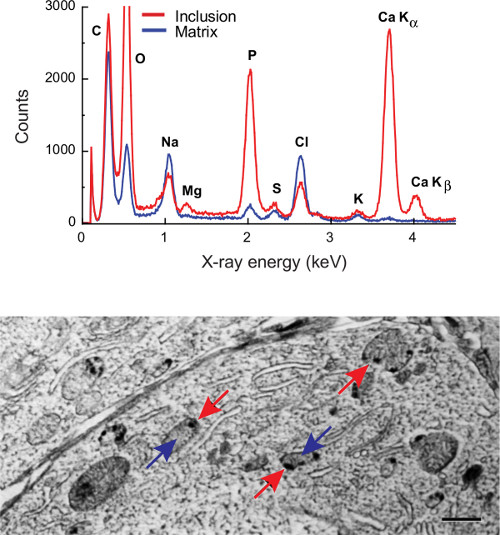
An EDX spectrum (Figure 2, inset) is essentially an x-y plot of the X-ray intensity vs. energy. Spectra contain qualitative and quantitative information about the elemental composition of the analyzed volume, in that the "characteristic" energy of a peak identifies the element giving rise to that peak while the intensity reflects the amount of that element. The characteristic peaks ride on top of a slowly varying background, the "continuum". (The legend to Figure 2 further discusses salient details of EDX spectra.) The energy of the peak manifolds for the entire periodic table are defined by the well known electronic structure of elements, thus all EDX software links to a database that automatically identifies components of an analyte. In a physiological context, elements of general interest that are well suited to EDX analysis include Na (Kα at 1.04 keV), P (2.01 keV), K (3.31 keV), and Ca (3.69 keV).
- Take advantage of available software database and peak matching routines to identify major elements in the spectra. In a biological specimen expect to find peaks for Na, Mg, P, S, Cl, K, and Ca. (CAUTION: The last two elements overlap!)
Quantitative analysis
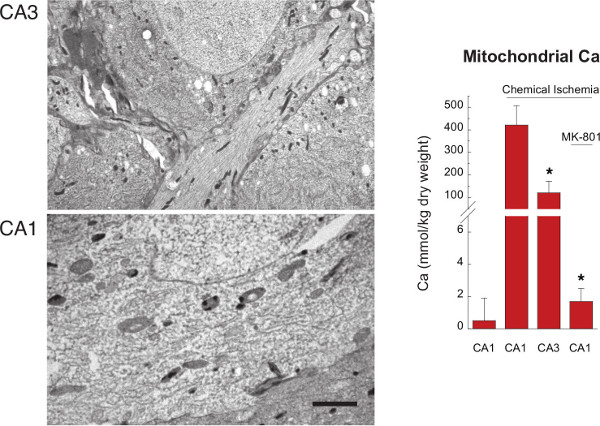
Quantitative analysis of EDX spectra consists of extracting the integrated area of identified peaks and converting this value to a concentration. For biological analysis, the established approach is the Hall peak/continuum method4,7,10, which takes advantage of the fact that the intensity of the continuum (defined above and in Figure 2) is proportional to the dry mass of the analyzed volume. Thus, the ratio of peak area/continuum area, when compared to the same ratio in spectra of standards of known composition, specifies the concentration in the targeted cellular compartment. Note that this approach provides concentrations in units of moles per weight, typically expressed as mmol/kg dry weight. This unit is unusual and for interpretation may require additional conversion to, for example, mmol/L wet weight or mmol/mg protein, as described elsewhere4,10.
- Extract peak areas (and error estimates) of biological elements between Z = 10-20 (0.5-4.0 keV), i.e. Na, Mg, P, S, Cl, K, and Ca, using one of the fitting routines built into analysis software (see Table 1, especially footnote 4). This lab uses Simplex or multiple-least squares fitting. Note that this fitting requires careful attention to resolving the overlap between K and Ca peaks (see Figure 2).
- Integrate the continuum between 1.45-1.61 keV. Alternative interference-free regions can be used.
- Calculate peak/continuum ratios and then concentrations by comparison to standards. Propagate errors throughout the analysis.
- Use standard statistical software and formulas to estimate averages that reflect biological variability.