Drosophila S2 cells attached on the ConA-coated coverslip spread into a flat round “pancake” shape (Figures 1A and 1C). However, when S2 cells plated on coverslips in the presence of CytoD and allowed to spread for 2-3 hr, they form long thin processes filled by parallel microtubules (Figures 1B and 1D). These microtubules have uniform polarity with plus-ends out7. Time-lapse fluorescence imaging of Drosophila S2 stably expressing mCherry-tubulin shows that processes are very dynamic, especially during the first 60 min after attachment (Figure 2). After 1 hr incubation, their growth slows down and 2-3 hr incubation ensures that majority of cells have fully grown processes. Here we show two representative examples of organelle transport along microtubules in S2 cells, lysosomes (labeled with LysoTracker, 200 nM; Figures 3A-B) and peroxisomes (labeled with peroxisome-targeted GFP, GFP-SKL)7 (stable transfection with pAC-GFP-SKL; Figures 3D-E). Time-lapse images of the cells were taken every 1 sec for 61 frames using a Nikon TU-2000 inverted microscope equipped with a Perfect Focus system (Nikon) and Coolsnap CCD camera (Roper Scientific) driven by Metamorph software. A 100-W halogen light source was used for fluorescence excitation to minimize photobleaching and phototoxicity. Each of the 61 frames in sequence was color coded according to the bar at upper right and images were superimposed to generate color-coded tracks. Thus moving particles generated rainbow tracks, while stationary particles appeared white10. ImageJ plug-in Temporal Color-Code (http://fiji.sc/Temporal-Color_Code) was used for this processing. For quantitative analysis of organelle movement we used DiaTrack software (Semasopht Inc.), we only select organelles that are localized in processes because they can be easily tracked and the polarity of microtubule tracks is known. Figures 3C and 3F show typical distributions of retrograde and anterograde velocities obtained by lysosomes and peroxisomes tracking, respectively.
Similar techniques are used in our lab for analysis of organelle transport in primary cultured neurons. Neurons are a predominant cell type in mixed cultures obtained from stage 9-11 embryos. After overnight culturing they are easy to recognize by characteristic cell shape with neurites that are over 50 µm long (Figure 4A). These cells are also positive for the pan-neuronal marker, Elav21 (Figures 4B and 4B'). Processes in the neurons are filled with microtubules (Figures 4B and 4B"). We can track organelle transport along the neurites using the techniques described above for S2 cells. Mitochondria can be labeled with mitochondrial GFP (Mito-GFP) under control of a motor neuron-specific promoter D4222 (Figures 5A and 5A'), and peroxisomes can be marked by injection of DNA encoding a peroxisome-targeted mCherry7, into syncytial blastoderm stage embryos (Figures 5B and 5B').
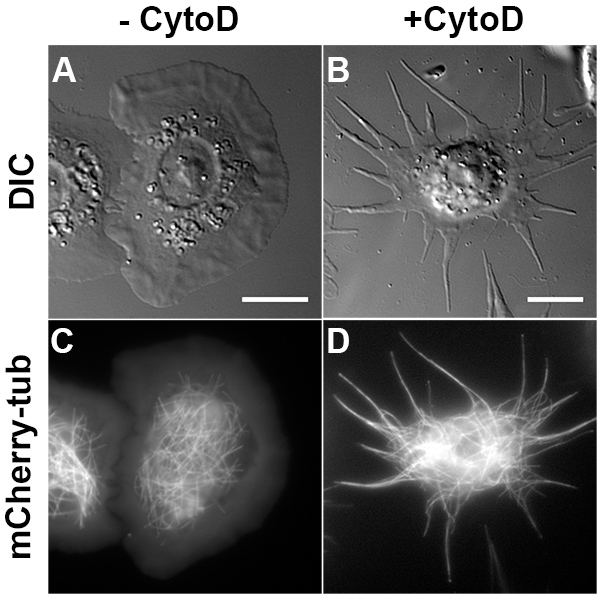
Figure 1. Drosophila S2 process formation induced by CytoD. (A, C) Drosophila S2 cells attach and spread into flatten and round shapes when plated in ConA-precoated coverslips. (B, D) S2 cells plated in the presence of 2.5 µm CytoD form long processes filled with microtubules after 3 hr incubation. A-B and C-D are transmitted light and fluorescent mCherry-tagged tubulin images, respectively. Scale bar, 10 µm. Click here to view larger image.

Figure 2. Time lapse of processes growth in a Drosophila S2 cell. Fluorescence time-lapse of a Drosophila S2 cell expressing mCherry-tubulin in the presence of 2.5 µM CytoD. The numbers indicate the time after the cell attach to the coverslip. Scale bar, 5 µm. Click here to view larger image.
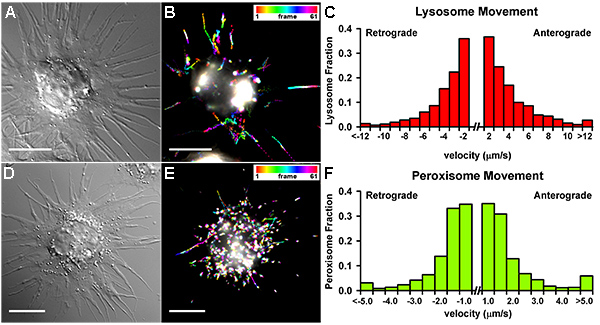
Figure 3. Organelle transport in Drosophila S2 cells. (A-C) Lysosome movement in Drosophila S2 cell. A representative 1 min-lysosome transport (labeled with 200 nM Lysotracker) in one cell (A, DIC image) represented by the artificial Temporal-Code (created by FIJI) (B). Velocities obtained from the analysis of lysosome movement tracked in processes (DiaTrack; 663 particles in 23 cells) (C). (D-F) Peroxisome movement in Drosophila S2 cell. A representative 1 min-peroxisome transport (labeled with pMT-GFP-SKL) in one cell (D, DIC image) represented by the artificial Temporal-Code (created by FIJI) (E). Velocities obtained from the analysis of peroxisome movement tracked in processes (DiaTrack; 228 particles in 20 cells) (F). Note that lysosomes move with longer and faster trajectories than peroxisomes. Scale bar, 5 µm. Click here to view larger image.
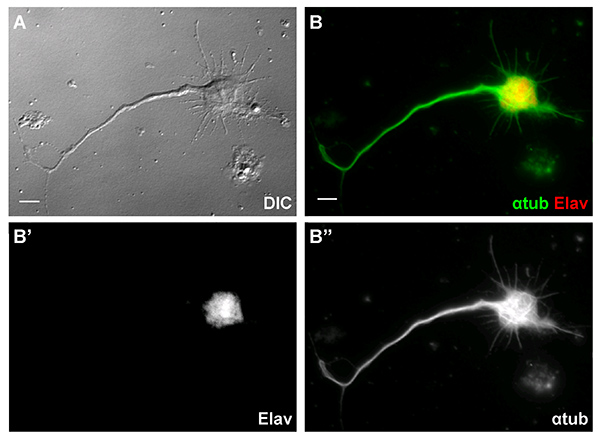
Figure 4. Primary Drosophila neurons from overnight culture. (A) An overnight cultured Drosophila neuron has characteristic neuronal morphology with long neurites. (B) The neuron expressed a pan-neuronal marker, Elav (red in B and white in B', DSHB, 7E8A10, 1:100) and with long neurites filled with microtubules (green in B and white in B'', DM1α, 1:1000). Scale Bar, 5 µm. Click here to view larger image.
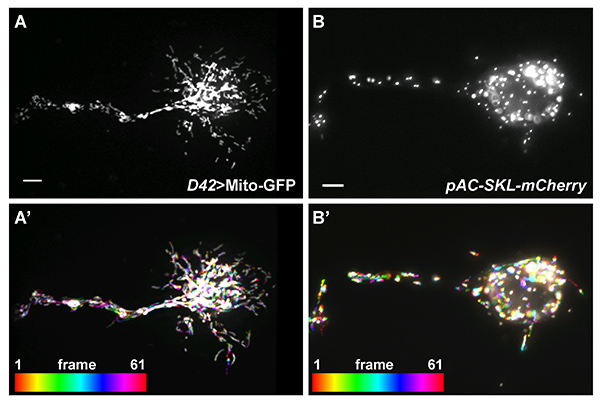
Figure 5. Organelle transport in cultured Drosophila neurons. (A) Mitochondria in a cultured Drosophila neuron marked by Mito-GFP under control of a motor neuron-specific promoter, D42. (A') 5 min-mitochondrial movement in (A) is represented by the artificial Temporal-Color code (created by FIJI). (B) Peroxisomes in a cultured Drosophila neuron marked by pAC-SKL-mCherry, injected into early syncytial blastoderm stage embryos. (B') 2 min-peroxisome movement in (B) is represented by the artificial Temporal-Color code (created by FIJI). Scale bar, 5 µm. Click here to view larger image.
| Organelle | Description |
| Peroxisomes | Peroxisomal targeting signal 1 tripeptide (SKL) tagged with a FP25 |
| Mitochondria | Mitochondrial targeting sequence of cytochrome c oxidase subunit VIII tagged with a FP26 |
| Mitochondria | Mitotracker (Dye which accumulates in active mitochondria)27 |
| Lysosomes | Lysotracker (Dye which accumulates in cellular compartments with low internal pH)28 |
Table 1. Most common organelle labeling strategies.
This table summarizes the most common organelle labeling strategies in tissue culture cells, including labeling of peroxisomes, mitochondria, and lysosomes.
