Analysis of RNA Processing Reactions Using Cell Free Systems: 3′ End Cleavage of Pre-mRNA Substrates in vitro
Summary
RNA polymerase II synthesizes a precursor RNA that extends beyond the 3′ end of the mature mRNA. The end of the mature RNA is generated cotranscriptionally, at a site dictated by RNA sequences, via the endonuclease activity of the cleavage complex. Here, we detail the method to study cleavage reactions in vitro.
Abstract
The 3’ end of mammalian mRNAs is not formed by abrupt termination of transcription by RNA polymerase II (RNPII). Instead, RNPII synthesizes precursor mRNA beyond the end of mature RNAs, and an active process of endonuclease activity is required at a specific site. Cleavage of the precursor RNA normally occurs 10-30 nt downstream from the consensus polyA site (AAUAAA) after the CA dinucleotides. Proteins from the cleavage complex, a multifactorial protein complex of approximately 800 kDa, accomplish this specific nuclease activity. Specific RNA sequences upstream and downstream of the polyA site control the recruitment of the cleavage complex. Immediately after cleavage, pre-mRNAs are polyadenylated by the polyA polymerase (PAP) to produce mature stable RNA messages.
Processing of the 3’ end of an RNA transcript may be studied using cellular nuclear extracts with specific radiolabeled RNA substrates. In sum, a long 32P-labeled uncleaved precursor RNA is incubated with nuclear extracts in vitro, and cleavage is assessed by gel electrophoresis and autoradiography. When proper cleavage occurs, a shorter 5’ cleaved product is detected and quantified. Here, we describe the cleavage assay in detail using, as an example, the 3’ end processing of HIV-1 mRNAs.
Introduction
The biosynthesis of most mature eukaryotic message RNAs (mRNAs) requires several post-transcriptional modifications such as capping, splicing and polyadenylation. These modifications are generally coupled to ensure correct processing1 and strongly increase the stability of the mRNA.
The 3’ end formation of mammalian pre-mRNAs is generated by endonucleolytic cleavage of the nascent RNA followed by addition of adenylate residues to the 5’ cleaved product by poly(A) polymerase (PAP)2-4. In mammals, cleavage is accomplished by a multicomponent protein complex, of approximately 800 kDa, which assembles on specific pre-RNA sequences. The poly(A) signal sequence, a highly conserved canonical hexanucleotide sequence AAUAAA, directs the cleavage site at approximately 10-30 nt downstream. This site is specifically recognized by the cleavage and polyadenylation specificity factor (CPSF) and the 73 kD subunit of CSPF contains the endonuclease activity. The cleavage stimulation factor (CstF) binds a more degenerate GU- or U- rich element sequence downstream of the poly(A) site. Also required for cleavage is the mammalian cleavage factor I (CFIm), and the mammalian cleavage factor II (CFII). CFIm binds the specific UGUA(N) sites in upstream sequence elements (USEs) that have been defined for a number of genes and seem to be involved in important physiological processes5-8.
In vitro, RNA processing reactions are commonly analyzed by the use of radiolabeled RNA substrates9-12. These may be synthesized by run-off transcription from the bacteriophage promoter T7 or SP6. When studying a polyadenylation site that has not been characterized before, it is necessary to use genomic DNA rather than cDNA to generate the RNA substrate, as important downstream sequences might not be present in cDNA. Design substrates to include at least 150 nt upstream and 50 nt downstream from the cleavage site/end on the mature mRNA. The cleavage product migrates faster than the substrate; however, because other fragments may be generated by non-specific nuclease action, the specificity of the reaction has to be verified by its dependence on the correct processing signal sequences. Therefore, RNA substrates with a point mutation in the AAUAAA sequence (e.g. AAGAAA) serve as a negative control for the cleavage reaction.
Given that a small amount of radiolabeled RNA is used for the cleavage reactions, RNases present in high abundance in most nuclear extracts can be problematic, and limit the choice of the starting material for extract preparation. HeLa cells tend to contain low levels of endogenous RNases, and thus perform well in these assays.
The endonucleolytic cleavage of the RNA substrates in vivo and in vitro is immediately followed by the poly(A) addition, thus the cleaved intermediate is not present in detectable quantities. Therefore, to study either a specific RNA sequence or proteins involved in a cleavage reaction, experiments are done in conditions that prevent polyadenylation from occurring. There is no dependence of cleavage on polyadenylation, or vice versa, so one can stop polyadenylation without harming the cleavage reaction. Thus, ATP is replaced with a chain terminating analogue that lacks the 3’ hydroxyl group so that only a single nucleotide can be incorporated at the poly(A) site and just cleaved RNA can be detected.
Given the complexity and high degree of particularity of this type of assay, we describe a detailed video protocol to study endonucleolytic cleavage by the cleavage/Poly(A) machinery of mRNA precursors in vitro. We describe how to prepare competent nuclear extracts, generate radiolabeled RNA substrates, perform the cleavage reaction, and analyze and interpret the resulting products. Figure 1 shows an example of substrate RNAs encoding for the 3’ end of HIV-1 pre-mRNAs to be used for a cleavage assay. The 3‘ end of the HIV RNA genome is composed of many important regulatory sequences such as the poly(A) site, a G+U rich region, and the USE element, which are all necessary for efficient maturation of the viral mRNA transcripts13. In this example we would expect the input RNA substrate to be 338 nt and upon cleavage 237 nt. If polyadenylation was allowed to occur, a smear of products would be observed between 237 and 437 nt.
Protocol
1. Adaptation of Adherent Cells into Suspension
(This is an optional step. Suspension cells generally make better nuclear extracts, however cells grown in plates may also be used.)
- Adapt adherent cells for growth in suspension using Joklik's modified MEM supplemented with 5-10% newborn calf serum or fetal bovine serum and 1% L-glutamine: penicillin-streptomycin. Propagate cells in spinner flasks with a filter cap at 37 °C with 8% CO2.
- When adapting cells, start with 100 ml in a 500 ml spinner flask. Maintain a minimum 0.3 x 106 cells/ml and replace the medium every 1-2 days until the proper growth rate is achieved (typically 2-6 weeks). Once adapted, HeLa cells in suspension should double approximately every 24 hr. NOTE: During the adaption phase, it will take some time for the cells to return to their normal doubling rate.
- Maintain the cells at a density of 0.5-0.8 x 106 cells/ml in the desired final volume (typically 1-10 L)
- Cells are typically harvested at mid log phase (about 0.5-0.65 x 106 cells/ml and >90% viable). Use the appropriate sized spinner flask for the desired amount of culture media.
2. Nuclear Extracts
- Buffer and Reagent Preparation
NOTE: Extractions are performed following a modified Dignam et al. (1983)14 nuclear extract procedure.- Prepare 1X phosphate buffered saline, buffer A, buffer C, and buffer D (Table 1) the day before proceeding with extractions and store at 4 °C.
NOTE: Higher yields are generally achieved with freshly prepared buffer; however, the buffer is stable for several weeks. It is important that all the buffers, apparatuses and equipment are pre-cooled and kept cold for the entirety of the extractions. Use a cold room for as many steps possible and/or keep everything on ice. Add DTT and PMSF immediately before use.
- Prepare 1X phosphate buffered saline, buffer A, buffer C, and buffer D (Table 1) the day before proceeding with extractions and store at 4 °C.
- Collecting and Washing Cells
- Pour cells into four 250 ml conical bottles and spin at 500 x g for 5 min at 4 °C in a centrifuge with a swinging or a fixed angle rotor. Decant supernatants and add another 250 ml cell culture medium to each bottle. Repeat spins until all the cells have been pelleted.
- Resuspend combined final pellets in a total of 250 ml of ice-cold PBS and pool into one 250 ml conical bottle. Spin the cells at 400 x g for 10 min at 4 °C in a swinging bucket centrifuge.
- Pour off the supernatant and resuspend the cell pellet in ice-cold PBS so the total volume does NOT exceed 50 ml. Loosen the pellet via pipetting and transfer to a 50 ml conical centrifuge tube.
- Spin the cells again at 500 x g for 6 min at 4 °C in a swinging bucket centrifuge. Pour off supernatant and estimate and note the cell pellet volume (CPV) as accurately as possible. Use water in a separate matching tube, for comparison, if the pellet is lower than the volume markings on the tube.
- Cell Lysis
- Add DTT to buffer A. Resuspend the pellet in buffer A using 5 volumes of the estimated CPV. Pipette to break up the pellet and incubate on ice for 10 min.
- Spin the cells at 931 x g for 10 min at 4 °C in a swinging bucket centrifuge.
- Carefully aspirate the supernatant instead of decanting. The pellet volume should have increased roughly two fold in size due to swelling of the cells.
- Add 1 CPV of buffer A to the cells. Gently break up the pellet and remove a small aliquot (about 50 µl) to a microfuge tube on ice. Transfer the resuspended cells into an appropriately sized Dounce homogenizer (usually 15 ml size) chilled on ice.
- Lyse the cells with 12-15 slow but steady strokes of a type B (tight) pestle. Remove a small aliquot of the lysate and examine under a microscope for complete lysis (intact small dark nuclei compared to swollen cells) by trypan blue staining. Intact cells will be clear). Continue homogenation until 90% lysis is achieved and stop here, over douncing may negatively impact quality of nuclear extracts.
- Transfer lysate into ultraclear ultracentrifuge tubes and note the total volume. Balance by weighing the tubes and spin at 1,069 x g for 10 min at 4 °C.
NOTE: The ultracentrifuge is used at this step despite the low spin speed because the same tube will be spun at higher speed in the next step. This spin will yield a fairly tight pellet covering the bottom of the tube with cloudier supernatant above, which may be collected and used for cytosolic S100 extracts. If there is a visible lipid layer on top of the tube, it can be removed. - Carefully remove the supernatant with a pipette and transfer it into another tube. Avoid disturbing the nuclear pellet. NOTE: It will be important to know the final volume of supernatant in order to determine packed nuclear volume (PNV).
- Preparation of the Nuclear Extract
- Once the supernatant has been carefully removed, respin the crude nuclear extract pellet at 25,453 x g for 15 min at 4 °C in the same tube.
- Remove the remaining supernatant (it should be a small amount of clear liquid) and add it to the previous supernatant collection. Measure the final volume of the supernatant. Calculate the PNV by subtracting this volume from the total volume of the original lysate previously noted. Alternatively, if the pellet is large enough, its volume can be estimated by comparing with similar volume of water in an identical tube.
- Add 1 PNV volume of buffer C (roughly 1 ml/ml of cells). Carefully dislodge the pellet with a pipette tip and transfer into an appropriately sized Dounce homogenizer. If using the same homogenizer as before, prerinse the homogenizer with sterile deionized water.
- Resuspend the pellet with ~5-10 strokes via a Dounce homogenizer using a tight pestle.
- Transfer the homogenized nuclear lysate into a chilled tube and mix by inversion for 30 min at 4 °C. If the volume is large enough, agitate using a magnetic stir plate and spin bar.
- Transfer the lysate into a new ultraclear centrifuge tube and spin at 25,453 x g for 30 min at 4 °C. The pellet should be compact. About 5 min before the spin is complete, hydrate the dialysis cassettes or tubing that have an appropriate molecular weight cut off (MWCO) (e.g. 7,000 kDa).
- Remove the supernatant (nuclear extract) from the tube into a chilled conical tube using a transfer pipette then proceed with dialysis.
- Dialysis
- Inject the extracts into hydrated dialysis cassettes as per manufacturer’s instructions and dialyze the nuclear extracts in 500 ml of Buffer D50 for 1 hr at 4 °C with stirring.
- Replace the buffer with 500 ml fresh, ice-cold Buffer D50 and dialyze for an additional 4 hr at 4 °C. This 4 hr step may be split into two 2 hr changes instead.
- Remove the extracts from the dialysis cassette and transfer to chilled tubes. Spin the extracts for 5 min at 1,500 x g at 4 °C.
- Aliquot the extracts into chilled, Eppendorf tubes and snap freeze in liquid nitrogen (50-100 µl per aliquot). Store at -80 °C for future use.
3. RNA Probe Synthesis
- Template Preparation
NOTE: Templates used for transcription may be PCR fragments or plasmids that contain a T7 or SP6 promoter upstream and immediately adjacent to the desired probe sequence. Plasmid templates must be linearized downstream of the template sequence of interest. We noted higher yield from PCR templates.- The T7/SP6 promoter can be inserted during the PCR reaction. Design the sense primer with the T7/SP6 promoter upstream of the target sequence.
- Amplify the desired sequence in 2-3 reactions per template via PCR per manufacturer’s instruction using a high-fidelity polymerase.
- Combine equivalent reactions and purify by gel extraction.
- Sequence the PCR products to verify template accuracy. (Optional)
- RNA Transcription with 32P Labeling
- Perform in vitro transcription from 1 µg of template with a commercially available kit that is compatible with the promoter (SP6 or T7) with the following modifications. Use 1 μl of fresh 3,000 Ci/mmol [α-32P] UTP, 0.5 μl of 10 mM cold UTP, 0.1 µl of 10 mM GTP, and 0.9 μl of 10 mM m7G(5’)ppp(5’)G RNA cap in a total final volume of 20 µl.
NOTE: The G-cap improves stability of the RNA transcripts in nuclear extracts (cap:GTP) 10:1 ratio. The cold UTP is added to prevent the 32P-UTP from being the limiting reagent. - Synthesize an RNA size ladder as denoted above without the G cap. Commercial templates are available for generating the RNA size marker.
- Incubate the transcription reactions at 37 °C for 1 hr (for SP6 use 40 °C). Once completed, perform DNase digestion of the template for 15 min at 37 °C.
- Following digestion, add 1 μl of 0.5 M EDTA and 5 μl of loading buffer II to the RNA substrates. Add 20 μl loading buffer II to the marker.
- Heat the RNA marker and probes at 95 °C for 3 min then place on ice.
- Perform in vitro transcription from 1 µg of template with a commercially available kit that is compatible with the promoter (SP6 or T7) with the following modifications. Use 1 μl of fresh 3,000 Ci/mmol [α-32P] UTP, 0.5 μl of 10 mM cold UTP, 0.1 µl of 10 mM GTP, and 0.9 μl of 10 mM m7G(5’)ppp(5’)G RNA cap in a total final volume of 20 µl.
- Polyacrylamide Gel Casting/Probe Electrophoresis
- Prepare 1 L of 6% polyacrylamide gel (PAGE) mix and 400 ml of 10% PAGE. To make 6%, mix 150 ml of 40% 19:1 acrylamide:bis-acrylamide with 100 ml of 10X Tris/Borate/EDTA (TBE) and 460 g urea. Bring up to 800 ml with sterile deionized water and stir on a hotplate while applying low heat (around 50-80 °C). Complete to 1 L with deionized water.
NOTE: The reaction is endothermic. Heating promotes the solubilization of the urea. Remove from heat as soon as the urea is dissolved and bring the solution to 1 L with deionized water. Alternatively stir at room temperature overnight; too much heat can initiate polymerization. - Protect the PAGE mix from light with aluminum foil and store at room temperature. The PAGE mixes can be prepared ahead of time and are stable for several months.
- Prepare 400 ml of 10% PAGE mix by combining 100 ml of the 40% 19:1 acrylamide:bis-acrylamide, 40 ml of 10x TBE and 184 g urea. After the urea dissolves, bring up to 400 ml with deionized water.
Alternatively, prepare a 20% 19:1 acrylamide solution and a 0% acrylamide solution with buffer and urea only; these can then be mixed in any proportion to give any percentage between 0 and 20%; urea is always slow to dissolve. - Prepare 10-50 ml of 10% ammonium persulfate (APS) solution in sterile deionized water depending on the number of gels to be cast.
- Wash 20 cm x 22 cm glass gel plates with 70% ethanol (EtOH). Dry with large lint free wipes.
- Wash the regular plate with 1 ml of 5 N sodium hydroxide (NaOH) and then rewash with 70% EtOH. Coat the notched plate with gel repel siliconizing solution by wiping the plate with a saturated small Kimwipe.
- Assemble the plates by placing the regular plate on an empty pipette tip box or a piece of Styrofoam to elevate it from the table with the cleaned side facing up. Lay 0.4 mm spacers on the edge of each long side.
- Carefully place the notched plate over the spacers so that the coated side will be on the inside of the gel. Use a razor or other thin instrument to shift the spacers for a flush alignment at the bottom and sides of the gel plates then secure with binder clips on each side of the gel plates over the spacers.
- Prepare 30 ml of 8% PAGE mix per gel by mixing 15 ml of 10% and 6% mixes each in a 50 ml tube. Quickly add 300 μl of 10% APS followed by 30 μl of N,N,N’,N’-tetra-methylethylenediamine (TEMED) to the 8% gel mix.
NOTE: Appropriate gel percentages for individual probe sizes should be selected by the experimenter. - Cap and invert 2x and pour the solution into the notched area of the gel plates. Be sure to slightly elevate the plates by hand and maintain an even pour until the PAGE mix starts to drip out of the bottom.
- Lower the plates back to level and immediately insert the large 8-well rectangle comb for 0.4 mm x 20 cm gel. Small bubbles near the bottom will not affect the running, but there should be no bubbles in the wells.
NOTE: Place paper towels on the table below the bottom and top of the gel to capture any spills during pouring. - Allow the PAGE mix to polymerize for at least 30 min.
- Transfer the polymerized gel to the cold room and assemble the electrophoresis apparatus. Add cold TBE buffer to the fill lines on the upper and lower chambers.
- Remove the comb and use a razor to remove any debris. Set the gel in the apparatus and use a binder clip to hold the plate to the seal. Add cold TBE to the top chamber until it flows into the plates.
- Wash the wells with a 30 ml syringe/21 G 1-1/2” needle filled with cold TBE just prior to loading the labeled RNA substrates. Place an aluminum plate to the back of the glass to aid in even distribution of heat while running.
- Load 3 μl of the marker and the entire 25 μl transcription reaction of each RNA substrate in separate wells. Leave a well between similarly sized transcripts to prevent cross-contamination. Bromophenol blue and xylene cyanol dyes in the loading buffer can be used to estimate size migration on the gel.
- Run the gel at a constant wattage appropriate for the size of the RNA probes (e.g. 25 watts for 2 hr for probes of 600 nt).
- Prepare 1 L of 6% polyacrylamide gel (PAGE) mix and 400 ml of 10% PAGE. To make 6%, mix 150 ml of 40% 19:1 acrylamide:bis-acrylamide with 100 ml of 10X Tris/Borate/EDTA (TBE) and 460 g urea. Bring up to 800 ml with sterile deionized water and stir on a hotplate while applying low heat (around 50-80 °C). Complete to 1 L with deionized water.
- Labeled RNA Extraction and Purification
- Following electrophoresis, carefully drain the top chamber of the gel apparatus. Most of the radioactivity will be in the gel and the bottom chamber; however, the upper buffer may contain some radioactive material.
- Use a tray to transport the gel if moving to another radioactive material work area. Carefully remove the spacers by pulling the protruding piece sideways away from the gel.
- Gently separate the plates. The gel should stick to the plate that was not siliconized. Place a piece of plastic wrap over the gel and glass plate.
- Place a glogos autorad marker sticker on the plastic wrap over an area of the gel that is clear of any samples.
- In a dark room, place a piece of autoradiography film over the entire plate and expose for at least 1 min. Develop the film.
- Place the developed film on a light surface. Then place the plastic side of the gel face down on the film and align the autorad marker from the gel to the exposed marker on the film. Once aligned, use a black marker to mark the location of the desired bands on the glass.
NOTE: Alternatively, align gels/film in the following way to cut out RNA bands. In the dark room, place the wrapped gel on a table, place the film on top of it and place a cassette (or book, etc.) on top of the piece of film. Flick on the light switch in the room briefly, then turn if off. This will 'preflash' the film generating an outline of all the wells on the film due to the pressure/protection from light that the cassette on top provides. This makes it easy to align the film on the gel and cut out the band without using fluorescent markers. - Carefully remove the plastic wrap and use a fresh scalpel or razor for each RNA substrate and excise the bands from the gel and place into separate 1.7 ml microfuge tubes. Use a nutator for gentle agitations during elution.
- Add 500 μl of RNA elution buffer (0.5 M ammonium acetate, 10 mM MgCl2, 1 mM EDTA, 0.2% SDS) to each tube. Briefly centrifuge to submerge the gel fragment. Incubate at room temperature in the dark for 3-16 hr depending on probe size.
- Transfer the entire 500 μl of eluted material into a new tube. Avoid transferring any gel pieces as they will interfere with the purification. Add 1 μl of glycogen (20 mg/ml) along with 500 μl of cold, acidic phenol-chloroform (for RNA).
- Briefly vortex, the solution should appear cloudy. Spin at room temperature at top speed (~16,000 x g) for 5-6 min.
- Carefully transfer the top (aqueous) layer to new tubes collecting as much as possible while avoiding any debris from the interphase. Add an equal amount of chloroform to the transferred aqueous layer. Repeat step 3.4.10 and transfer the top (aqueous) layer to a new tube. Add 1 ml of ice-cold 95-100% EtOH to the collected material. Incubate at -80 °C for 10 min. NOTE: The RNA may be stored overnight if desired.
- Pellet the RNA for 30 min at 16,000 x g at 4 °C. Carefully pour off the supernatant into a radioactive waste container and add 1 ml of ice-cold 70% molecular grade EtOH to each pellet.
- Pellet the RNA at 16,000 x g for 15 min at 4 °C, pour off supernatant, spin again for 1 min and remove any residual EtOH via pipette and leave at room temperature with the caps open to allow for evaporation of any remaining EtOH.
- Resuspend the pellets in 20-30 μl of RNase-free water. Labeled RNA may be stored at -80 °C.
NOTE: A radioactive survey meter should be used throughout the entire extraction procedure to verify that the labeled RNA has not been lost during any step.
- Quantitation of Labeled RNA for a pre-mRNA 3’ mRNA Cleavage Reaction
- Approximate the molarity of the cleavage substrates using the following procedure.
- Take, for example, 1 μl of 3,000 Ci/mmol [α-32P] UTP and 10 mCi/ml. (On the label reference date, the chemical concentration of this stock solution will be 3.3 µM UTP. Before that date it is higher and one half-life (~13 days) after the date it is ~1.6 mM, etc.) The labeled UTP contribution to the final reaction concentration is typically negligible when a final cold UTP concentration above the UTP Km is used. The T7 RNAP Km for UTP is 114 mM 15.
- Dilute the 1 μl in 39 μl of water. Then add 1 μl of diluted [α-32P] UTP to an appropriate volume of scintillation fluid in a scintillation vial. (NOTE: Scintillation fluid is not actually needed for this specific activity of 32P as Cerenkov counting will give reliable cpm values too. Check the scintillation counter manual to choose the correct channel for Cerenkov counting and count a 1 ml volume for 1 min).
- Dilute 1 μl of each labeled RNA substrate into separate vials with an equal amount of scintillation fluid used in the previous step. Make sure to have a vial with only scintillation fluid to calculate background. Quantitate radioactivity in a scintillation counter.
- Multiply the counts per min (cpm) for the [α-32P] UTP sample by 40 to factor in the initial dilution and subtract the background obtained by the scintillation fluid alone. Multiply the counts for each RNA probe by the amount of RNase-free water (20-30 μl) used to resuspend the RNA probes. This gives the total cpm of the purified RNA.
Example: Max cpm = (cpm [α-32P] UTP – background) x 40. Labeled RNA Sample 1 = 20,000 cpm x 20 = 400,000 cpm (if 20 μl used for resuspension) - If an amount other than 1 μl of [α-32P] UTP was used to make the transcripts, then factor this volume into the final cpm of [α-32P] UTP by multiplying the cpm for the [α-32P] UTP sample by the amount used.
Example: 2 μl of [α-32P] UTP is used to make the transcripts. The calculated cpm for 1 µl [α-32P] UTP is 500,000 cpm. Multiply 500,000 * 2 = 1,000,000 cpm - Divide the calculated labeled RNA cpm by the calculated cpm for [α-32P] UTP alone. This will roughly estimate the percentage of labeled UTP incorporated.
Example: 400,000 / 1,000,000 = 0.40 or 40% incorporated - Since an enzyme cannot distinguish between isotopes, the same percentage of labeled and unlabeled UTP is incorporated into the RNA during transcription. Calculate the amount of cold UTP in moles that is added to the transcription reaction (0.5 μl of a 10 mM solution =5.0 x 10-9 moles of UTP).
- Multiply the moles UTP in the reaction by the percent incorporated (known from the hot UTP counting above), then divide that by the number of uridines in the RNA sequence.
Example: 5.0 x 10-9 moles of UTP x 0.40 = 2 x 10-9 moles UTP
2 x 10-9 moles UTP / 73 U per transcript = 2.7 x 10-11 moles of RNA - Convert moles of RNA to molarity by dividing the volume of the recovered RNA resuspension solution.
Example: 2.7 x 10-11 moles of RNA / 20 μl = 1,370 nM RNA. For T7 RNAP transcripts made from about 1 µg linearized plasmid and resuspended in 20 µl water, values of 200 to 1,400 nM are typical.
NOTE: The molar concentration of RNA is used to ensure that the final pre-mRNA substrate concentration in the in vitro cleavage reaction is less than 5 nM per cleavage reaction. Cleavage efficiency can decrease if the RNA concentration is raised significantly above 5 nM.
- Approximate the molarity of the cleavage substrates using the following procedure.
4. 3’ mRNA Cleavage Reaction
- Cleavage Reaction Reagent Preparation
- Prepare all the reagents needed for the cleavage reaction. Make 10% polyvinyl alcohol (PVA) by boiling water and slowly add the correct amount of PVA powder. 10% PVA will be viscous and takes time to dissolve. Store at room temperature or -20 °C.
- Prepare 1 M creatine phosphate (CP) and store at -80 °C. It is important to use 1 M CP that is less than 4 months old.
- Prepare 1 M DTT and store at -20 °C. Make a fresh 0.2 M DTT dilution from the 1 M DTT just prior to doing the cleavage reaction.
- Prepare ETS (cleavage stop solution): 10 mM Tris pH 7.8, 10 mM EDTA, 0.5% SDS. Store at room temperature.
- Cleavage Reaction
- Heat the labeled RNA to 80 °C for 2 min then place on ice.
- Gently vortex and then spin the 10% PVA at top speed for 2-3 min to remove bubbles.
- Master mix 1: For a single reaction, combine 3.125 μl 10% PVA, 0.625 μl 1 M CP, 0.25 μl 0.5 M tRNA, 0.125 μl 100 mM dATP, 0.13 μl 40 U/μl RNase inhibitor, 0.25 μl 0.1 M EDTA pH 8, 0.1 μl 0.2 M DTT, and 0.645 μl RNase-free water. Dispense 5.25 μl master mix 1 per tube on ice.
- Master mix 2: Use 6.25 μl/reaction of nuclear extracts at concentrations higher than 2 mg/ml. Nuclear extracts may be diluted with Buffer D50 if needed.
- Combine the 6.25 μl of nuclear extract with the previously dispensed 5.25 μl of master mix 1. Add 1 μl of labeled RNA substrate that is diluted to 50 nM. Alternatively, the RNA may be included in the Master mix 1 provided that it is added after the RNase inhibitor, DTT and tRNA. NOTE: Use <5 nM of the labeled RNA substrate.
- Gently vortex and quick spin each sample to remove bubbles. Incubate at 30 °C for up to 2 hr, but a time course should be performed for optimal results. Longer incubations might increase background degradation.
- Proteinase K Digestion and Purification
- Remove the samples from heat and add 182.5 μl of ETS stop solution to each reaction. Add 5 μl of 20 mg/ml proteinase K and incubate samples at 37 °C for 10 min.
- Extract the RNA by adding 1 volume (200 μl) of acid phenol-chloroform, 1/10th volume (20 μl) of 3 M sodium acetate, and 1 μl of 20 mg/ml glycogen. Proceed with extractions as described in sections 3.4.9-3.4.14.
- Once the RNA extraction is completed, be sure to remove all residual EtOH. Resuspend the pellet in 8 μl of loading buffer (90% formamide, 10 mM EDTA, with Bromophenol Blue (BFB) (and or Xylene Cyanol Blue)). The samples may be stored at -80 °C or proceed with gel electrophoresis.
- Cleavage Reaction Electrophoresis
- Prepare a 6% acrylamide gel as previously stated in sections 3.3.1-3.3.17 with a few modifications. Use 20 cm x 42 cm sequencing gel plates with 2 binder clips on each long side. Use a 32-well comb and prepare 40 ml of the gel mix.
- Load the gel with 3 μl of marker and 5 μl of each sample. Optimize the electrophoresis settings according to pre-cleavage and cleaved product sizes.
- When electrophoresis is completed, follow sections 3.4.1-3.4.3 to disassemble the gel. Instead of plastic wrap, cut a piece of filter paper to the size of the gel plate and carefully place it on one side of the exposed gel.
- Press the filter paper firmly onto the gel, then flip over the plate so that the filter paper is on the bottom and the glass is on top. Firmly press the glass down on the table to ensure that the gel is gripping the filter paper evenly.
- Slowly peel the filter paper, which should have the gel attached to it, from one of the short ends until all of the filter paper with gel attached is removed from the glass plate.
- Cover the exposed gel side with plastic wrap. Tape the plastic wrap to the filter paper.
- Place the gel on a gel dryer with the filter side facing the vacuum side. Dry for 15 min or until the gel is completely dry.
NOTE: If a gel dryer is not available, X-ray film may be used to visualize the RNA bands. Exposure should be carried out in a light-tight cassette with intensification screen at -80 °C. Exposure time is determined empirically. Caution: low percent acrylamide gels may crack upon freezing. - The gel may now be used to expose film or be used with a phospho-imager. Exposure time depends on strength of the labeled RNA. Initially a 24 hr exposure may be used. If using film, expose at -80 °C with an intensification screen and allow to warm to room temperature prior to developing.
- Measure the ratio between cleaved and un-cleaved RNA in each sample to quantitate cleavage efficiency.
Representative Results
Representative results of a cleavage assay of the RNA poly(A) sites of HIV-1 (Figure 2). We can observe the uncleaved RNA substrate, which is the slowest migrating band at the top of the gel. The specific cleaved product is the most intense shorter fragment band that runs faster in the gel at the expected size, and is specifically absent from the cleavage assay of the mutpoly(A) control that contains a point mutation in the polyA sequence (mutPolyA) RNA substrate. Degradation products of the input substrate may sometimes be observed. Quantification of the cleavage activity may be determined by densitometric plot of the ratio of uncleaved to cleaved RNA normalized to 100% of uncleaved RNA.
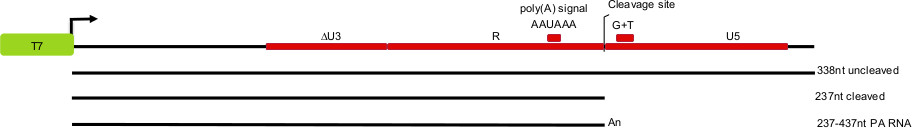
Figure 1. Schematic depiction of the HIV-pre-mRNA 3’ end processing substrates, and expected products of in vitro cleavage and polyadenylation reactions. The cleavage site, the dinucleotide –CA-, lies 25 nt downstream from the polyA site AAUAAA. LTR regions: U3 (unique 3’ sequence), R (repeated sequence), and U5 (unique 5’ sequence).
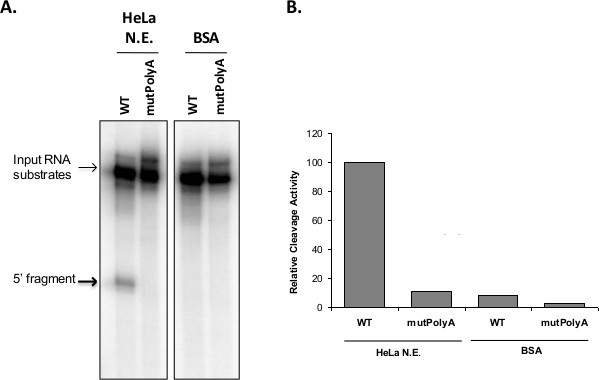
Figure 2. (A) Poly(A) site cleavage. 32P-labeled RNAs containing the poly(A) of HIV-1 and a point mutation of the poly(A) signal that abolishes cleavage (mutPolyA) were incubated in nuclear extracts of HeLa-S3 cells or BSA as a negative control. Bolded arrow indicates 5’ cleaved product. The 3’ fragment often runs off the gel or is degraded and not always visible. (B) Densitometric plot of the cleavage activity expressed as a ratio of uncleaved to cleaved RNA normalized to 100% of uncleaved RNA.
| Buffer A (100 ml) | 10 mM Tris (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT). | Add DTT just prior to using buffer A during the extraction. Add 1 μl of 1 M DTT per 1 ml of buffer A. |
| Buffer C (50 ml) | 20 mM Tris (pH 7.9), 25% (v/v) glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride (PMSF). | Add a protease inhibitor cocktail tablet the morning of the extractions. |
| Buffer D50 (2-4 L) | 20 mM Tris (pH7.9), 20% (v/v) glycerol, 0.2 mM EDTA, 0.5 mM DTT, 50 mM ammonium sulfate. | HEPES-KOH may be substituted for Tris in buffer A, C and D. |
| RNA Elution Buffer | 0.5 M amonium acetate, 10 mM MgCl2, 1 mM EDTA, 0.2% sodium dodecyl sulfate (SDS) | Store at room temperature. |
| Stop Buffer (ETS) | 10 mM Tris (pH8.0), 10 mM EDTA, 0.5% SDS | Store at room temperature. |
| Cleavage Loading Buffer | 90% formamide, 5 mM EDTA pH8, 0.025% Bromophenol Blue | Store at -20 °C. |
Table 1. Composition of buffers.
Discussion
The in vitro pre-mRNA 3’ cleavage reaction, carried out in HeLa cell nuclear extracts or with cleavage factors fractionated from these extracts, has enabled identification of the core cleavage factors and their main complexes16-21. Many more proteins associated with these factors have recently been identified22, and the in vitro reaction may continue to shed light on how these proteins contribute to the reaction. Perhaps because the reaction in vivo appears to be cotranscriptional23, or because some contributing factors may be lost during extraction and dialysis, the efficiency is often poor, and rarely is more than 30% cleavage is achieved. In addition, the reaction efficiency may suddenly drop from day to day for no apparent reason. Therefore, it is important to appreciate which steps are the most critical, especially when attempting to adapt the reaction to modified or virally-infected HeLa cells, to other cell types or to new substrates.
Without doubt, the quality of the extract and the freshness of the in vitro transcribed RNA are paramount, as is the need to work under scrupulously RNase-free conditions. The health of the cells at the time they are harvested for extraction is also important. The cells should appear healthy, they should be doubling in less than 24 hr, they should be in or approaching mid log phase, and there should be few dead cells or other unexplained debris. Obviously, the cells should not be contaminated by mycoplasma or other bacteria. The substrate should not be made with too high a specific activity, as this can lead to apparent degradation.
When a large enough sequence on either side of the cleavage site is considered, and alternative polyadenylation is accounted for, the number of different poly(A) sequence signals in the human genome undoubtedly exceeds the number of genes24. As there is variability in the “strength” of different signals that have been tested, when adapting the procedure to a new cell type, it is best to begin with one of the stronger polyadenylation signals, such as the often-used Simian virus 40 (SV40) late poly(A) signal25. The combination of a weak poly(A) signal sequence with cells other than standard HeLa cells can prove frustrating, and it is better first to use a strong substrate with new cells, or unmodified HeLa cells with a potentially weak polyA substrate.
Even after so many years of successful use, there are still minor mysteries associated with the in vitro 3’ cleavage reaction. For example, why is creatine phosphate needed? It has been demonstrated that the original reason for including it – to regenerate the ATP pool – is not valid26. Interestingly, it can be omitted with only partial loss of activity when nuclear extracts are used, but becomes progressively more necessary as the extract is fractionated into partially purified cleavage factors that are used to reconstitute the reaction. In fact, phosphocholine, another small molecule with a phosphate group and a positively charged amine, but likely having no in vivo role in the reaction, has been found to be more effective than creatine phosphate, at least in the reconstituted reaction27. PVA is another ingredient whose requirement is not fully explained. It is assumed to be a crowding agent, leading to an effective increase in cleavage factor concentration, but other crowding agents, like polyethylene glycol, do not work nearly as well. Understanding these factors might lead to improved efficiencies, enabling the extension of the method to less efficient cell types and RNA substrates, and might yield clues to how the various factors work.
Declarações
The authors have nothing to disclose.
Acknowledgements
S.V. is grateful for the funding support from the NIH K22AI077353 and the Landenberger Foundation. K.R. gratefully acknowledges funding from the NIH (5SC1GM083754).
Materials
| 1L Celstir Flask | Wheaton | 356884 | Different sizes available |
| 4 Position Slow Speed Stirrer | VWR | 12621-076 | |
| Swinging Bucket Centrifuge | Beckman Coulter | Allegra x-15R with SX4500 Rotor | |
| Ultra Centrifuge | Beckman Coulter | Optima L-100 XP Ultra with SW41 Rotor | |
| Table Top Centrifuge 5417R | Eppendorf | Refridgerated | |
| Thermomixer incubator | Eppendorf | ||
| 250ml Conicle Tubes | Corning | 430776 | |
| 50ml Conicle Tubes | BD Falcon | 352098 | |
| Ultra-clear Centrifuge Tubes | Beckman Coulter | 344059 | |
| JOKLIK Modified MEM | Lonza | 04-719Q | |
| Fetal Bovine Serum | Atlas | F-0500-A | Heat inactivated |
| L-Glut:Pen:Strep | Gemini Bio-Products | 400-110 | |
| 1M Tris-HCl pH 8.0 | Mediatech | 46-031-CM | |
| Magnesium Chloride | Fisher | BP214-500 | |
| Potassium Chloride | MP Biomedicals | 194844 | |
| HEPES | Fisher | BP310-500 | |
| DTT | Alexis Biomedicals | 280-001-G-010 | |
| Glycerol | Fisher | BP229-4 | |
| 5M Sodium Chloride Solution | Mediatech | 46-032-CV | |
| EDTA 0.5M Solution | Sigma-Aldrich | E7889-100ml | |
| EDTA | Fisher | BP120-500 | |
| PMSF | Thermo Scientific | 36978 | |
| Ammonium Sulfate | Fisher | A702-500 | |
| cOmplete EDTA-Free Protease inhibitor cocktail tablets | Roche | 04 693 132 001 | |
| Slide-A-Lyzer Dialysis Cassettes Kit | Thermo Scientific | 66372 | MWCO 7000 0.5ml-3ml |
| 15ml Dounce Tissue grinder set | Sigma-Aldrich | D9938-1SET | Different sizes available |
| Expand High Fidelity PCR Kit | Roche | 11 732 650 001 | |
| 10mM dNTP Mix | Invitrogen | Y02256 | 10mM each nucleotide |
| MaxiScript SP6/T7 Kit | Ambion | AM1322 | |
| m7G(5')ppp(5') G RNA Cap | New England Biolabs | S1404S | |
| Century Marker Template Plus | Ambion | AM7782 | |
| Easytides UTP [alpha-32p]-250uCi | Perkin Elmer | BLU507H250UC | |
| Gel loading buffer II | Ambion | 8546G | |
| DEPC treated water | Ambion | AM9906 | |
| 10x TAE | Fisher | BP13354 | |
| 10x TBE | Ameresco Life Sciences | 0658-4L | |
| 10x PBS | Fisher | BP399-20 | |
| Urea | Fisher | BP169-212 | |
| Ammonium Persulfate | Bio-Rad | 161-0700 | |
| TEMED | Fisher | BP150-20 | |
| Ammonium Acetate | Fisher | A637-500 | |
| 40% 19:1 Acrylamide:Bis-acrylamide | Bio-Rad | 161-0144 | |
| Glycogen | Roche | 10 901 393 001 | |
| 100% Absolute Ethanol 200 Proof | Acros | 61509-0040 | |
| Acid Phenol-Chloroform | Ambion | 9720 | For RNA |
| Scintilation Fluid | Fisher | SX18-4 | |
| Rnase Inhibitor | Promega | N261B | |
| Poly (Vinyl alcohol) PVA | Sigma-Aldrich | P8136-250G | |
| Creatine Phosphate | Calbiochem | 2380 | |
| 100mM dATP | Fisher | BP2560-4 | |
| SDS | Acros | 23042-5000 | |
| Proteinase K | Fisher | BP1700-100 | |
| Adjustable Sequencing Unit | Sigma-Aldrich | Z351881-1EA | |
| Binder Clips | Office Depot | 838-056 | |
| 20cmx42cm glass plates | Sigma-Aldrich | Z352543 | 1SET |
| 20cmx22cm glass plates | Sigma-Aldrich | Z35252-7 | 1SET |
| 20cmx42cm Aluminum Cooling Plates | Sigma-Aldrich | Z352667 | 1EA |
| 0.4mmx22cm Spacers | Sigma-Aldrich | Z35230-6 | 1SET |
| 0.4mmx42cm Spacers | Sigma-Aldrich | Z352314-1 | 1SET |
| 8-well Comb | Sigma-Aldrich | Z35195-4 | 1EA |
| 16-well Comb | Sigma-Aldrich | Z351962 | 1EA |
| 32-well Comb | Sigma-Aldrich | Z351970 | 1EA |
| Gel Repel Coating | C.B.S. Scientific | SGR-0401 | or SGR-0101 for individual bottle |
| Gel Loading Tips | Rainin | GT-10-4 | 0.1-10uL |
| Sequencing PowerPac HV | Bio-Rad | PowerPac HV | 5000V/500mA/400W |
| Gel Dryer Model 583 | Bio-Rad | Model 583 | |
| Hydrotech Vacuum Pump for gel dryer | Bio-Rad | ||
| Glogos II Glow-in-the-dark Markers | Agilent | 420201 | |
| Film 8×10 | Midsci | BX810 | |
| Film 14×17 | Phenix | F-BX1417 | |
| Autoradiography Cassette | Fisher | FBCA 1417 | 8×10 size available |
Referências
- Maniatis, T., Reed, R. An extensive network of coupling among gene expression machines. Nature. 416, 499-506 (2002).
- Wahle, E., Ruegsegger, U. 3′-End processing of pre-mRNA in eukaryotes. FEMS microbiology reviews. 23, 277-295 (1999).
- Colgan, D. F., Manley, J. L. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 11, 2755-2766 (1997).
- Matoulkova, E., Michalova, E., Vojtesek, B., Hrstka, R. The role of the 3′ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA biology. 9, 563-576 (2012).
- Danckwardt, S., et al. The prothrombin 3’end formation signal reveals a unique architecture that is sensitive to thrombophilic gain-of-function mutations. Blood. 104, 428-435 (2004).
- Moreira, A., et al. The upstream sequence element of the C2 complement poly(A) signal activates mRNA 3′ end formation by two distinct mechanisms. Genes Dev. 12, 2522-2534 (1998).
- Hall-Pogar, T., Zhang, H., Tian, B., Lutz, C. S. Alternative polyadenylation of cyclooxygenase-2. Nucleic Acids Res. 33, 2565-2579 (2005).
- Brackenridge, S., Proudfoot, N. J. Recruitment of a basal polyadenylation factor by the upstream sequence element of the human lamin B2 polyadenylation signal. Molecular and Cellular Biology. 20, 2660-2669 (2000).
- Moore, C. L., Sharp, P. A. Accurate cleavage and polyadenylation of exogenous RNA substrate. Cell. 41, 845-855 (1985).
- Gilmartin, G. M. . mRNA formation and function. , 79-98 (1997).
- Wahle, E., Keller, W. . RNA Processing. Vol. II. , 1-34 (1994).
- Chabot, B. . RNA Processing Vol I. 1, 1-29 (1994).
- Valente, S. T., et al. HIV-1 mRNA 3′ end processing is distinctively regulated by eIF3f, CDK11, and splice factor 9G8. Mol Cell. 36, 279-289 (2009).
- Dignam, J. D., Lebovitz, R. M., Roeder, R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475-1489 (1983).
- Aurup, H., Williams, D. M., Eckstein, F. 2′-Fluoro- and 2′-amino-2′-deoxynucleoside 5′-triphosphates as substrates for T7 RNA polymerase. Bioquímica. 31, 9636-9641 (1992).
- Keller, W., Bienroth, S., Lang, K. M., Christofori, G. Cleavage and polyadenylation factor CPF specifically interacts with the pre-mRNA 3′ processing signal AAUAAA. EMBO J. 10, 4241-4249 (1991).
- Takagaki, Y., Ryner, L. C., Manley, J. L. Four factors are required for 3′-end cleavage of pre-mRNAs. Genes Dev. 3, 1711-1724 (1989).
- Takagaki, Y., et al. A multisubunit factor, CstF, is required for polyadenylation of mammalian pre-mRNAs. Genes Dev. 4, 2112-2120 (1990).
- Ruegsegger, U., Blank, D., Keller, W. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol Cell. 1, 243-253 (1998).
- Vries, H., et al. Human pre-mRNA cleavage factor II(m) contains homologs of yeast proteins and bridges two other cleavage factors. EMBO J. 19, 5895-5904 (2000).
- Gilmartin, G. M., Nevins, J. R. An ordered pathway of assembly of components required for polyadenylation site recognition and processing. Genes Dev. 3, 2180-2190 (1989).
- Shi, Y., et al. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell. 33, 365-376 (2009).
- Bentley, D. L. Rules of engagement: co-transcriptional recruitment of pre-mRNA processing factors. Curr Opin Cell Biol. 17, 251-256 (2005).
- Tian, B., Manley, J. L. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 38, 312-320 (2013).
- Ryner, L. C., Manley, J. L. Requirements for accurate and efficient mRNA 3′ end cleavage and polyadenylation of a simian virus 40 early pre-RNA in vitro. Molecular and Cellular Biology. 7, 495-503 (1987).
- Hirose, Y., Manley, J. L. Creatine phosphate, not ATP, is required for 3′ end cleavage of mammalian pre-mRNA in vitro. J Biol Chem. 272, 29636-29642 (1997).
- Ryan, K., Khleborodova, A., Pan, J., Ryan, X. P. Small molecule activators of pre-mRNA 3′ cleavage. RNA. 15, 483-492 (2009).
