G protein-coupled receptors (GPCRs) constitute one of the largest and most diverse families among all cell surface proteins. Their presence in vertebrates, invertebrates, plants, yeast, and slime mold, as well as in Protozoa and the earliest diploblastic Metazoa indicates that GPCRs are among the oldest molecules linked with signal transduction1. Their natural activating ligands comprise a wide diversity of external stimuli including peptides, biogenic amines, odorants, glycoproteins, and photons2. As such, these receptor-ligand signaling systems are involved in a great variety of physiological processes. The broad functional spectrum makes them ideally suited for the development of therapeutic drugs that cover a wide range of human diseases. About 50-60% of the current drug targets are represented by GPCRs3,4. Besides their great importance in the pharmaceutical industry, GPCRs are also in the spotlight for the development of a new generation of species-specific insecticides5,6 and pesticides in general. Because the natural ligands of many GPCRs are still unidentified, they are classified as orphan GPCRs. The deorphanization of these receptors will improve the understanding of their physiological roles in organisms and may uncover putative targets for new drug applications7.
Since the genomic era, the reverse pharmacology strategy is widely applied for the deorphanization of GPCRs8. The approach implies that an orphan receptor is used as a ‘hook’ to ‘fish out’ its activating ligand from a biological extract or from a library of synthetic compounds. The GPCR of interest is therefore cloned and subsequently transfected in a cellular expression system. In the most commonly used methods, receptor activation is determined by measuring changes in the concentration of second messenger molecules9. The main receptor screening assays rely on calcium-sensitive bioluminescent proteins (e.g., aequorin)10 or fluorescent calcium indicators (e.g., Fluo-4)11. The fluorescence-based assays, in which receptor-expressing cells are loaded with a fluorescent calcium indicator prior to ligand screening, have the advantage that they allow high-throughput screening due to their ease of use, short reading time, and the flexibility of screening multiple orphan receptors on a single plate12.
Here, the fluorescence-based calcium mobilization assay is thoroughly described and illustrated by the deorphanization process of the Drosophila melanogaster short neuropeptide F (sNPF) receptor. This neuropeptidergic signaling system was originally characterized by Mertens et al. in 200213 with a calcium bioluminescence assay performed in Chinese hamster ovary (CHO) cells14 and by Feng et al. in 2003 with an electrophysiological assay using Xenopus oocytes15. The presence of the sNPF signaling system seems to be limited to the phylum of Arthropoda, where it is implicated in a wide range of processes including the regulation of feeding, growth, stress reactions, locomotion, and circadian rhythms16.
Research on neuropeptidergic signaling systems in insects may not only lead to new targets for the development of insecticides, but the knowledge of their functioning may also be extrapolated towards other organisms as many signaling systems have been generally well conserved throughout evolution17. In the last decade, great progress has been made in the deorphanization process of insect neuropeptide GPCRs. Despite these efforts, only a small number of receptors have been matched to their cognate ligand, and loads of sequence information for new orphan GPCRs has become available due to the booming of genomics18. The availability of medium/high-throughput screening approaches, like the fluorescence-based calcium mobilization assay that has proven to be a widely applied technique9,18, is therefore invaluable.
The fluorescence-based calcium mobilization assay as described here is performed in the human embryonic kidney 293T (HEK293T) cell line and uses a fluorescent probe to determine changes in intracellular calcium concentrations upon receptor activation. To guarantee high expression and translation levels of the receptor, a Kozak consensus sequence19 is added to the 5’ end of the receptor-coding sequence, which is subsequently cloned in an expression vector (e.g., pcDNA vector series for mammalian cell lines). As it is difficult to predict the endogenous G protein-coupling of an orphan GPCR based on sequence information alone, the second messenger molecules (e.g., calcium or cAMP) that are modulated after receptor activation often remain unknown prior to ligand identification. To circumvent this problem, promiscuous G proteins of the Gq family (e.g., murine Gα15 or human Gα16 [used here]) or chimeric G proteins (e.g., Gαqi5) that interact with most GPCRs and induce the release of calcium can be co-expressed20,21,22. Upon binding of the ligand to its receptor, the GPCR undergoes a conformational change that leads to the activation of specific intracellular pathways. The guanosine diphosphate (GDP) molecule, bound under resting conditions to the Gα16 subunit, will be replaced by a guanosine triphosphate (GTP) molecule. This provokes the dissociation of the heterotrimeric G protein in a Gα16 and Gβγ subunit. The Gα16 subunit activates phospholipase Cβ (PLCβ), which in turn hydrolyzes the membrane-bound phosphatidylinositol bisphosphate (PIP2) resulting in diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 will spread throughout the cytoplasm and activates IP3-dependent calcium channels present in the membrane of the endoplasmic reticulum, which induces the release of calcium into the cytoplasm.
The calcium release upon receptor activation occurs within seconds and can be detected by loading cells before the screening assay with a calcium-sensitive dye, like Fluo-4 acetoxymethyl (AM)11. The AM ester group enables the fluorophore to cross the cell membrane and is cleaved off by cytoplasmic esterases once inside the cell. Consequently, the negative charges of the fluorescent dye are unmasked, preventing it from diffusing out of the cell and allowing it to interact with calcium ions. The fluorescent signal of Fluo-4 is negligible in cells under resting conditions only containing calcium concentrations in the nanomolar range. However, when calcium is released upon receptor activation, the signal can increase concentration-dependently to more than a 100-fold, hereby ensuring a large signal-to-noise ratio. Fluo-4 also exhibits a large dynamic range for reporting [calcium] around a Kd(calcium) of 345 nM, making it suitable to measure physiologically relevant calcium changes in a wide range of cells. The excitation of Fluo-4 occurs at 488 nm and the emission fluorescence is measured at 525 nm11. Fluorimeters like the fluorescence imaging plate reader (FLIPR)23, the NOVOstar, or the FlexStation (station device)12 are medium/high-throughput systems that allow simultaneous compound addition and the detection of the Fluo-4 signal upon receptor activation for every well in an assay plate. The calcium mobilization assay described here relies on the station device 96-well microplate system.
The SoftMax Pro software (software) is used to operate the station device as well as for data analysis. The program immediately displays the results as graphs in 96-well format. Multiple wells can be selected simultaneously to compare the outcome of these wells on the same graph. The relative fluorescent unit (RFU) values of wells in each column are simultaneously measured for a period of two minutes, starting before the addition of compounds to the wells and continuing after measurement of the fluorescent signal following receptor activation. Typically, the trend of an agonist curve aligns with the baseline until an activating compound is added to the cells, resulting in a rapid increase of the fluorescent signal. The peak height is correlated with the final agonist concentration in the well. After the peak, the fluorescent signal slowly drops towards the baseline level. The RFU measurements can be converted into concentration-response curves to determine the EC50 value (half maximal effective concentration) of a ligand. In general, at least three independent screens, each including three replicas of a concentration series, should be performed to compose a reliable concentration-response curve.
It is recommended to include several positive and negative controls in the experimental design. First of all, a transfection control, i.e. the implementation of a receptor with a known ligand, should be tested. This allows verifying whether the transfection agent was operational. Incorporation of a control experiment with an agonist for an endogenous receptor of the cell line and a negative control (e.g., wash buffer) are also recommended to monitor the health and viability of the cells and to exclude the possibility that the wash buffer was contaminated with a factor that could elicit an auto-fluorescent response. Frequently used agonists are a peptide derived from the protease-activated receptor-1 (PAR1), which acts as a PAR1 selective agonist, or carbachol, which activates the acetylcholine receptor. Cells transfected with an empty expression vector should also be tested to exclude that active compounds interact with the cell’s endogenous receptors. Optimization of several parameters described in the protocol below may be required for different signaling systems. A schematic figure of the complete fluorescence-based calcium mobilization assay is depicted in Figure 1.
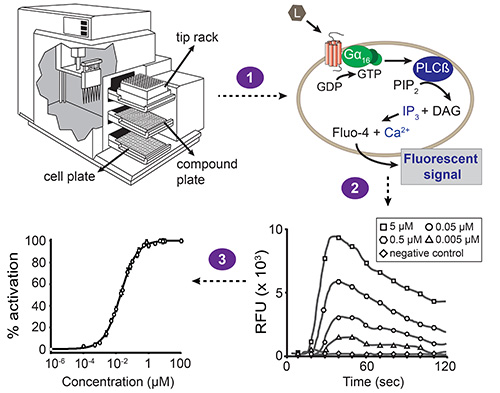
Figure 1. Overall scheme of the fluorescence-based calcium mobilization assay. Automated liquid handling and simultaneous fluorescence measurements are performed with the station device microplate reader, driven by the software. The station device contains three drawers: one for the cell plate, compound plate and tip rack. The build-in pipettor transfers compounds from one column of the compound plate to the corresponding column of the cell plate (step 1). Each well of the cell plate contains a monolayer of HEK293T cells that have been co-transfected with the GPCR of interest and the promiscuous Gα16 subunit. When a compound activates the receptor, the Gα16-bound GDP is replaced by GTP. The Gα16 subunit subsequently dissociates from the Gβγ complex and activates phospholipase Cβ (PLCβ), which in turn hydrolyzes phosphatidylinositol bisphosphate (PIP2) resulting in diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 activates IP3-dependent calcium channels present in the membrane of the endoplasmic reticulum, inducing the release of calcium into the cytoplasm. The interaction of calcium with Fluo-4 (with which the cells are loaded prior to compound addition) results in a fluorescent signal (step 2). The software presents the results as relative fluorescent unit (RFU) values in function of time, and peak heights correlate with the ligand concentration in a concentration-dependent manner. These data can then be converted into a concentration-response curve to determine the EC50 value of a ligand-receptor pair (step 3).
Concentration series ranging from 50 µM to 0.001 nM were tested for all four Drosophila sNPF peptides (Drome-sNPF-1: AQRSPSLRLRFamide, Drome-sNPF-2: SPSLRLRFamide, Drome-sNPF-3: PQRLRWamide, Drome-sNPF-4: PMRLRWamide) on HEK293T cells that transiently express the Drosophila sNPF receptor and the promiscuous Gα16 subunit. The GPCR was activated by all four peptides in final concentrations up to 0.1 nM, and receptor activation was concentration-dependent. Figure 2 depicts the graphs corresponding to one of three replicas of the Drome-sNPF-1 concentration series. The negative control (wash buffer) did not induce a fluorescent signal, while the positive control (PAR1 – 1 µM) elicited strong activation of the endogenous protease-activated receptor-1, leading to a high fluorescent signal (± 30,000 RFU). It should be noted that a deviation of the typical curve may indicate abnormalities. For example, an ongoing rise of the curve without returning to the baseline might be related to non-receptor-mediated signals, such as the presence of calcium ionophores, or a disrupted lipid bilayer causing calcium leaks.
The software allows entering formulas to determine the percentage of activation or the standard errors of the different concentrations among others. The resulting data are used to compose preliminary concentration-response curves to estimate the EC50 values, as shown for the four Drome-sNPF peptides in Figure 3. The curves of Figure 3 also include the data for the negative control where the concentration series of the Drome-sNPF peptides were tested on HEK293T cells transfected with an empty pcDNA3.1 vector. The results indicate that the addition of the peptides to these cells have no effect on endogenous receptors, but indeed activate the receptor of interest. The fluorescence-based calcium mobilization assay was then repeated three times independently. Based on the preliminary concentration-response curves of the initial screen, the tested concentrations were adapted to cover the dynamic range of the curve (Figure 4). The EC50 values of Drome-sNPF-1 (2.04 ± 1.48 nM [95% confidence interval]), Drome-sNPF-2 (5.89 ± 3.74 nM), Drome-sNPF-3 (5.55 ± 3.95 nM) and Drome-sNPF-4 (0.50 ± 1.01 nM) are similar, indicating that they are equally potent to activate the receptor.
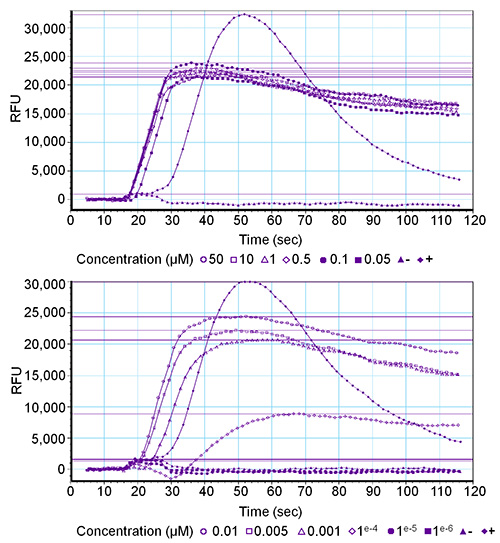
Figure 2. Graphical output of the software for a receptor-mediated fluorescence response of a concentration series. Twelve final concentrations (ranging from 50 µM to 0.001 nM) of the Drome-sNPF-1 peptide were tested on the Drome-sNPF receptor. Activation is expressed in relative fluorescent unit (RFU) values. The upper graph gives the results of the six highest concentrations and the lower graph of the six lowest concentrations. The positive control (+) is PAR1 (1 µM) and the negative control (-) is wash buffer.
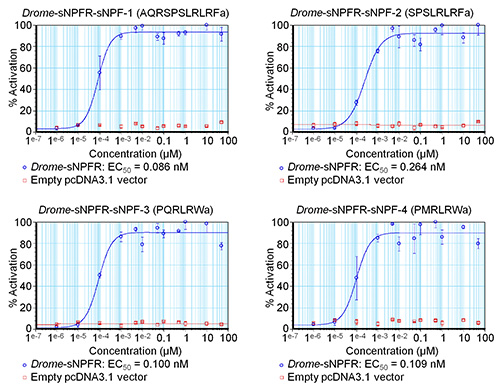
Figure 3. Preliminary concentration-response curves of Drosophila sNPF peptides determined by a single fluorescence-based calcium mobilization assay. The concentration-response curves of the Drosophila sNPF receptor transiently expressed in HEK293T cells for the four Drosophila sNPF peptides are shown in blue. For the negative controls (shown in red), peptides were tested on HEK293T cells transfected with an empty pcDNA3.1 vector. The fluorescent responses are shown as relative (%) to the highest value (100% activation). The curves are the result of one experiment in which each concentration series was measured in triplicate. The vertical bars represent standard errors of the mean (SEM), which sometimes are smaller than the used symbols (in that case, only the symbols are depicted).
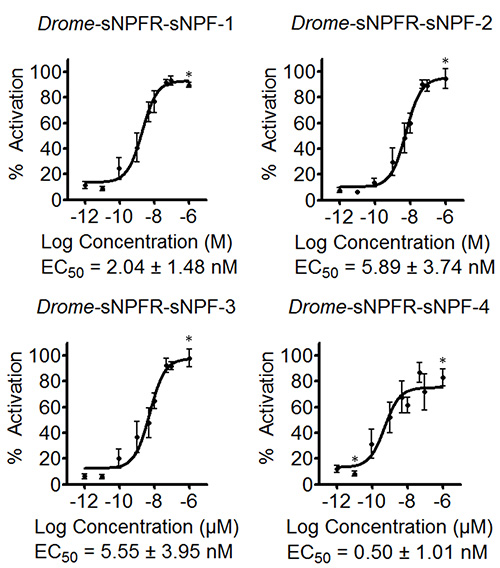
Figure 4. Concentration-response curves and corresponding EC50 values of Drosophila sNPF peptides determined by three independent fluorescence-based calcium mobilization assays. The concentration response curves of the Drosopihla sNPF receptor transiently expressed in HEK293T cells for the four Drosophila sNPF peptides are the result of three independent measurements each performed in triplicate (n ≥ 9). The fluorescent responses are shown as relative (%) to the highest value (100% activation). Asterisks indicate concentrations for which n ≤ 9. Error bars indicate SEM, which sometimes are smaller than the used symbols (in that case, only the symbols are depicted). EC50 values are shown with their 95% confidence intervals.
