A Method for Screening and Validation of Resistant Mutations Against Kinase Inhibitors
Summary
Emergence of genetic resistance against kinase inhibitor therapy poses significant challenge for effective cancer therapy. Identification and characterization of resistant mutations against a newly developed drug helps in better clinical management and next generation drug design. Here, we describe our protocol for in vitro screening and validation of resistant mutations.
Abstract
The discovery of BCR/ABL as a driver oncogene in chronic myeloid leukemia (CML) resulted in the development of Imatinib, which, in fact, demonstrated the potential of targeting the kinase in cancers by effectively treating the CML patients. This observation revolutionized drug development to target the oncogenic kinases implicated in various other malignancies, such as, EGFR, B-RAF, KIT and PDGFRs. However, one major drawback of anti-kinase therapies is the emergence of drug resistance mutations rendering the target to have reduced or lost affinity for the drug. Understanding the mechanisms employed by resistant variants not only helps in developing the next generation inhibitors but also gives impetus to clinical management using personalized medicine. We reported a retroviral vector based screening strategy to identify the spectrum of resistance conferring mutations in BCR/ABL, which has helped in developing the next generation BCR/ABL inhibitors. Using Ruxolitinib and JAK2 as a drug target pair, here we describe in vitro screening methods that utilizes the mouse BAF3 cells expressing the random mutation library of JAK2 kinase.
Introduction
Protein kinases are key regulatory enzymes of intracellular signal transduction pathways that seemingly modulate every cellular function. A proper control of kinase mediated signaling is crucial to homeostasis and development, which mostly relies on proper regulation of kinases, phosphatases and its degradation by UPS (ubiquitin proteasome system). Deregulated kinases are at the center stage of many cancers and implicated in host of human diseases 1. Human genome encodes more than 500 protein kinases that have been linked, directly or indirectly, to ~400 human diseases 2. These observations supported the concept for therapeutic targeting of kinases by small molecule inhibitors 3-5.
The demonstration of ABL kinase inhibitors, such as Imatinib, in the treatment of chronic myeloid leukemia (CML) provided the proof of concept for this approach6,7. This observation not only revolutionized the anti-kinase therapy but also enforced the idea to identify the genetic lesions in other neoplastic diseases for therapeutic targeting, which lead to discovery of oncogenic mutations in the JAK2 from polycythemia vera (PV) and patients with myeloproliferative neoplasms (MPN). This discovery generated great interest in treating MPNs by targeting JAK2 with small molecule kinase inhibitors. Now, almost a dozen of JAK2 inhibitors are in clinical trials and one of them has been approved recently for the treatment of myelofibrosis. While specific targeting of oncogenic kinases by small molecule inhibitors in cancers bring promising outcome, it also suffers from developing resistance to the treatment. In fact, so far, patients treated with kinase inhibitors, such as Imatinib, Gefitinib, Erlotinib and Dasatinib developed resistance mutations mostly by acquiring mutations in the kinase domain to which drug targets 8-10. Resistance as a result of gene mutation highlights the limitations of targeted monotherapy against the oncogenic kinases, and represents the next challenge in the development of ever more successful cancer chemotherapy. Mechanistic and functional consequences of drug resistance should provide a rationale for selection and design of complimentary compounds for drug development. Mutations identified via in vitro screens, have shown a high degree of correlation with those found in patients. Therefore, in vitro screening for mutations that confer drug-resistance for a given drug target pairs in clinical or preclinical development assists in identifying the resistance patterns that are likely to cause clinical relapse. The identification of these mutant forms is not only helpful in monitoring the patients for drug response and relapse but also essential for the design of more robust next generation inhibitors. For instance, development of next generation BCR/ABL inhibitors, Nilotinib and Ponatinib, were made possible because of greater mechanistic understandings gained from mutagenesis, structural, and functional studies.
Earlier, we have reported the results of our screen using random mutagenesis of BCR/ABL to reveal the spectrum of mutations conferring resistance to inhibitors such as Imatinib11,12, PD16632612, and AP2416313. The results not only identified the mutations conferring clinical resistance and disease relapse, but also provided the mechanistic understanding of drug resistance and principles governing the kinase function11,14. Here we provide additional methodological detail, using Ruxolitinib and JAK2 as a drug target pair, to enable a broader application of this screening strategy.
Protocol
NOTE: All procedures in this protocol were conducted according to the National Institute of Health guidelines for the ethical treatment and care of animals, and according to an approved IACUC animal use protocol.
1. Cell Line Maintenance
- Culture BAF3 cells in RPMI-1640 medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 units/ml and 100 µg/ml) and spent culture medium of Wehi Cells. Grow HEK293T cells in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 units/ml and 100 μg/ml). Maintain cells at 37 °C in a humidified atmosphere containing 5% CO2.
2. Plasmid Construction
- Clone the mouse JAK2 and its oncogenic isoform JAK2-V617F in pMSCV-puro-GW by LR clonase using standard recombination cloning procedure.
- For the construction of luciferase expression vector (pMSCV-Luc-Cherry-GW) used in in-vivo imaging, amplify luciferase from pLVX-Tet-ON vector by PCR using primers (LUC-SD/RP TTACAATTTGGACTTTCCG and LUC-SD/FP-CACCATGGAAGACGCCAAAAAC) followed with cloning in pENTR-SD-TOPO to generate pENTR-Luc, which is used to transfer the Luciferase gene in retroviral vector, pMSCV-Cherry-GW by recombination mediated cloning using LR clonase.
3. Preparation of Random Mutation Library, Screening and Identifying the Mutations
3.1) Random Mutagenesis
- Clone full length of JAK2 wild type and the V617F mutation into pMSCV-puro-GW retroviral vector using a commercial recombination cloning technology.
- Use 1 µg of JAK2-V617F plasmid DNA to transform, XL-1 Red, E. coli cells, which is defective in DNA repair mechanisms thus allowing them to incorporate random mutations during replication. More specifically, mix 50 – 100 ng of DNA (more than 100 ng of DNA decreases transformation efficiency) with 100 µl of competent cells in a pre-chilled polypropylene tube and incubated on ice for 30 min, gently swirling every 5 min. For a good library coverage, use four to six tubes of competent cells.
NOTE: More than 100 ng of DNA decreases transformation efficiency - Immerse the tube in a water bath at 42 °C for a heat shock of 45 sec and incubate on ice for 2 min. Next, add 1 ml of SOC medium (2.0% tryptone, 0.5% yeast extract, 0.05% NaCl, 10 mM MgCl2, 10 mM MgSO4, and 0.4% D-glucose). Incubate the tube at 37 °C while shaking at 225-250 rpm for 90 min.
- Plate cells from each tube on four 10 cm LB-agar plates containing 100 µg/ml ampicillin. Incubate the plates for 16 – 24 hr at 37 °C.
- Following visible colonies, collect them by scraping the plates with a sterile plate scraper. Pool cells from each plate and isolate plasmid DNA using a commercial plasmid extraction kit.
NOTE: Usually colonies are smaller and take 18-24 hr to be visible because these are slow growing bacterial strains. At this stage, assess the heterogeneity of mutations in the library by restriction digestion with a frequent cutter such as Sau3A1 or Taq1 or sequencing.
3.2) Production of Retroviral Supernatants and Transduction
- One day before transfection, plate 4 x 106 HEK 293T cells onto 100 cm dishes in DMEM containing 10% FCS, penicillin/streptomycin and 2 mM L-glutamine.
- Next day, replace the medium and transfect the cells with mutagenized pMSCV-JAK2-V617F library. For each 10 cm plate, mix 10 µg of DNA (5 µg of the plasmid library and 5 µg of retroviral packaging plasmid, pCLEco) with 40 µl of FuGENE to a total volume of 400 µl using serum free DMEM media. Incubate the DNA and lipid mix for 20 min at room temperature. After 20 min add the DNA-lipid complex drop wise on top of HEK293T cells.
- After six hr, change the transfection media with fresh media and incubate the plates for 48 hr at 37 °C for virus production. After 48 hr of incubation, collect viral supernatants filtered through a 0.45 µm acrodisc filter.
3.3) Selection of Resistant Clones In Vitro
- For drug resistant JAK2-V617F mutants, transduce 100 million BAF3 cell with 100 ml of virus supernatants having a viral titer of 0.5-1 x106. More specifically, mix 108 cells with 100 ml of virus supernatant to a total volume of 300 ml using RPMI media, containing 10% serum and 24 µg/ml of polyberene (final concentration of ployberene is 8 µg/ml). Distribute these cell mixers in multiple six well plates (4-5 ml per well).
- Centrifuge the plates at 1,250 x g for 90 min at 25 °C. After centrifugation, incubate the plates at 37 °C for 24 hr.
- Next day, pool the cells together and mix with Ruxolitinib and medium containing soft agar and plated in six well plates. For each drug concentration, mix 10 ml of transduced BAF3 cells (10-20 million cells) with the appropriate amount of Ruxolitinib in a 50 ml tube. Make up the volume to 40 ml using RPMI containing 20% serum. Add 10 ml of 1.2 % agar to the cells, mixcarefully and plate in six well plates.
- Incubate the plates at 37 °C for two weeks. Pick the resistant colonies using 0.2 ml pipette tips and sub-culture them individually in 24 well plates for 3-4 days.
- Harvest the resistant BAF3 cells by centrifugation at 450 x g for five min at room temperature. Isolate the genomic DNA using a commercial genomic DNA isolation kit. PCR amplify full length JAK2 cDNA from 100 ng of genomic DNA using primers (mJAK2FP-ATGGCCTGCCTTACAATGACAG and mJAK2RP-GGTTCTGGAAGTCTGCTTGG) and long-template expand high fidelity PCR systems.
- Separate the PCR products by agarose gel electrophoresis. Excise the 3.4 kb of DNA band representing JAK2 coding frame and isolate the DNA using a commercial gel extraction kit. Sequence the full length of cDNA using 8 internal primers (P1 – P8) spanning the coding sequence and analyze sequences using commercial software.
4. In Vitro Validation of Resistant Mutations
Many cell clones carry more than one mutation, To test the contribution of each mutation in resistance phenotype, generate selected variants by site-directed mutagenesis using pMSCV-JAK2V-617F plasmid as template.
- Perform site directed mutagenesis on pMSCV-JAK2-V617F plasmid using a commercial mutagenesis kit and oligonucleotides designed to create point mutations. Confirm the identities of mutant clones by sequencing.
- Transduce BAF3 cells with retroviruses made using mutant plasmids followed with selection using puromycin at 1µg/ml.
- Plate 104 BAF3-JAK2-V617F cells (50 µl of RPMI with 10% FCS) into each well of a 96 well plate. Separately add 50 µl of the ruxolitinib containing media to the wells across the plate (final concentration: 0, 1, 3, 5, 10, and 20 µM), and incubate the plates for 60 hr at 37 °C.
- Assess cell viability by adding 10 µl of WST-1 reagent to each well followed with incubation at 37°C for 2-4 hr. Record absorbance at A450 using a plate reader. Perform all assays in triplicate and plot averaged absorbance against INCB018424 concentrations. Perform a best-fit sigmoidal curve using a nonlinear curve fitting algorithm. Score the drug concentration resulting in 50% cell viability as the cellular IC50.
- Next, plate six million BAF3 cells expressing JAK2 variants in six well plates in RPMI medium containing 10% serum with increasing concentrations of ruxolitinib and incubated for 4–6 hr at 37 °C.
- Collect the cells by centrifugation at 450 x g for five min at 4 °C. Wash the cells one time with PBS, and lyse in 20 mM Tris-Cl (pH 7.5), 50 mM NaCl, 1% NP-40, 0.1% SDS, 5 mM EDTA, 1mM EGTA, 1 mM sodium fluoride, 2 mM Sodium Vanadate, and 5% Glycerol supplemented with protease inhibitor cocktail and phosphatase inhibitors. Sonicate the cell suspension for 1 min followed by addition of 5x gel loading buffer (350 mM Tris-HCl [pH 6.8], 500 mM DTT, 15% SDS, 10 mM Benzamidine, 5 mM EDTA, 5 mM EGTA, 5 mM Sodium Vanadate, Protease cocktail, 50% Glycerol, and 0.001% Bromophenol blue) and denaturation at 70 °C for 5–10 min.
- Resolve the proteins on a 8% SDS polyacrylamide gel under denaturing conditions. Perform immunoblotting with mouse monoclonal anti-phopsho STAT5. Strip the blots and reprobed with a rabbit polyclonal anti-SATA5 antibody. Visualize the bands using ECL reagents according to the supplier’s recommendation.
5. In Vivo Validation
- Transduce BAF3 cells expressing luciferase and cherry (transduced with retroviruses made from pMSCV-Luc-cherry GW) with viruses expressing JAK2-V617F and resistant variants. Select cells that survive puromycin selection for the expression of JAK2 and its resistance variants.
- Inject two million actively growing BAF3-Luc/cherry cells carrying JAK2-V617F and its resistant variants in 200 µl of PBS to 6 Balb/C mice via tail-vein injection.
- After three days of cell transplantations, inject the mice with Ruxolitinib (100 mg/kg) twice daily for two weeks.
- Perform luciferase-based bioluminescence imaging with an imaging system. .
- In brief, prepare luciferin solution by dissolving 300 mg to 25 ml of sterile deionized water, aliquot in smaller volumes and store.
NOTE: Store luciferin at -80 °C and protect from light until use. - Inject 250 µl to each mouse by IP to achieve 125 mg/kg in each mouse. Anesthetize the mice from same group together using inhaled isoflurane (1.5%-2.0%) in a sealed box. Apply vet ointment on the eye to prevent dryness.
NOTE: The time taken for mice to sleep (10-15 min) allowed luciferin activity to peak. Keep the time consistent between the groups to avoid variation.
- In brief, prepare luciferin solution by dissolving 300 mg to 25 ml of sterile deionized water, aliquot in smaller volumes and store.
- Transfer the mice immediately to imaging chamber stage and maintain anesthesia at 1.5-2% isofluorane during imaging procedures. Image mice with sequential 0.1, 0.5, 1, 5, 30, 60, and 300 sec exposure. Capture images and quantitate bioluminescence intensity using image acquisition and analysis software. Following imaging return the mice back to its cage.
- For, in vivo chimerism harvest total bonemarrow cells. Euthanize the mice with CO2 for 5 min followed with cervical dislocation. Harvest the bones and crush in 4 ml cold PBS to collect the bone marrow. Analyze the percent cherry positive cells under fluorescence microscope and quantify using FACS. Collect twenty thousand events from each sample.
Representative Results
Emergence of genetic mutations poses great challenge for the targeted anti kinase therapy. Mutational studies, besides providing mechanistic and functional insights that are instrumental in selection and design of next-generation drug development, also allows better clinical management and may in future be more helpful for personalized treatment. In this experiment, we show screening for ruxolitinib resistance mutations in JAK2-V617F kinase (Figure 1). We constructed pMSCV-JAK2-V617F-cherry.gateway vector by introducing the full-length mouse JAK2-V617FcDNA into pMSCV-Cherry-GW. It is recommended to use bacterial host (XL-1 red strain of E. coli) to develop random mutations over the most popular choice of method, which is PCR, due to its limitations associated with sequence bias and larger gene fragments are difficult to amplify. Randomly mutagenized DNA library was transfected to HEK293T cells for retrovirus production. These mutant viruses were used to, transduce the BAF3 cells that were selected for colony growth in soft-agar in the absence of IL-3 with either 1 or 5 µM of Ruxolitinib. Under these conditions, colonies arise only from cells carrying JAK2V-617F cDNAs that expresses functional and resistant variant of the kinase. After 10 – 14 days, well-separated individual colonies were picked, which varied in size, and expanded them in liquid culture. Genomic DNA was isolated from these cells. The provirus was recovered by direct rescue or by PCR. The recovered proviruses were sequenced to identify mutations (Figure 1). Sequence analyses were performed using DNASTAR package of Lasergene.
Because many cell clones carried more than one mutation, we strongly recommend validating these mutations in isolation by both in vitro and in vivo assays (Figures 2 and 3). To verify the activity of variants in isolation, selected variants were generated by site directed mutagenesis of the JAK2V-617F sequence. These variants were reintroduced into BAF3 cells, and measured for cell proliferation ability at different dosage of Ruxolitinib to evaluate IC50 (IC50 values, Figure 2A). Biochemical assays are performed for phosphotyrosine or phospho-STAT5 by immunoblot analysis on protein lysates of BAF3 cells that were incubated with increasing doses of Ruxolitinib to rule out any off target mediated resistance (Figure 2B). Mutants exhibiting enhanced IC50 values and persistent autophosphorylation at higher Ruxolitinib concentrations are thus confirmed to be drug resistant variants. Because many resistant mutations show variable dose response, therefore it is imperative to test whether these mutations will confer in vivo resistance as well. Usually, we recommend to test only 2 -3 different variants for in vivo experiments, as they are expensive and laborious. To validate in vivo resistance, BAF3 cells were engineered to express JAK2-V617F variants and Luciferase/cherry to enable in vivo tracking. Mice were injected with 1-2 million cherry positive cells (expressing JAK2 variants and luciferase), followed by Ruxolitinib injection for two weeks. After two weeks, mice were imaged for luciferase-catalyzed bioluminescence (Figure 3), and also for BAF3 chimerism in bonemarrow and peripheral blood by measuring the fluorescence of cherry positive cells (Figure 3). Mice expressing JAK2-V617F are sensitive to Ruxolitinib, while resistant variants show progressive increment in bioluminescence over the period of treatments, thus suggesting that BAF3 cells expressing JAK2-V617F variant is resistant to Ruxolitinib treatment in vivo.
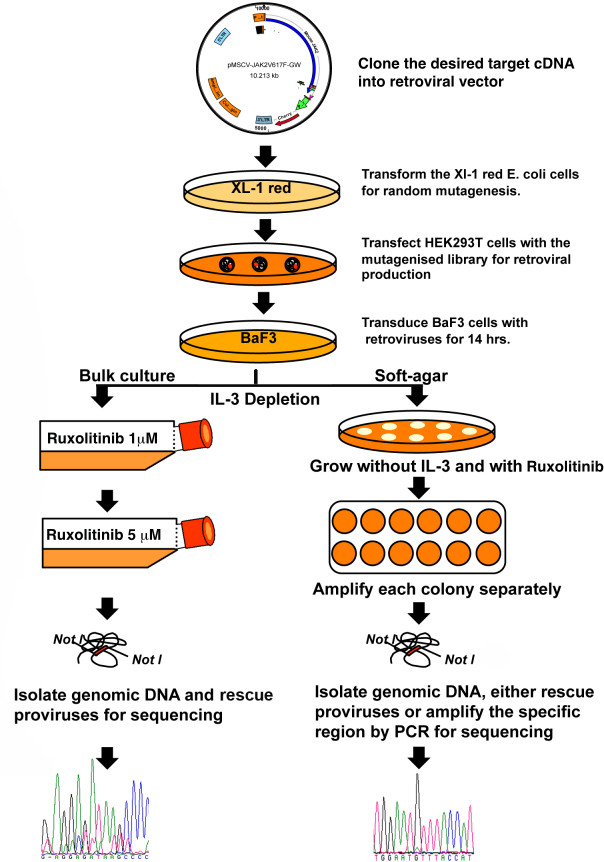
Figure 1: A scheme for screening the resistance mutations against kinase inhibitor Ruxolitinib.
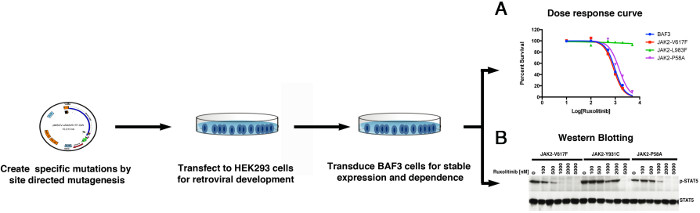
Figure 2: A scheme showing in vitro validation using dose dependent cell proliferation assays (A) and western blotting (B). Please click here to view a larger version of this figure.
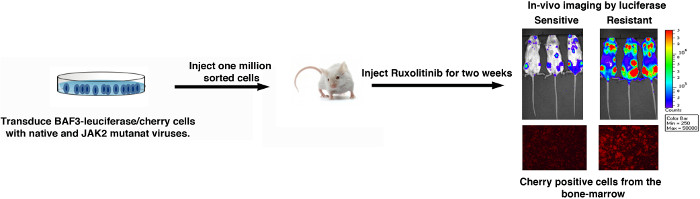
Figure 3: A schematic representation of in vivo validation using luciferase catalyzed bioluminescence measurement. Please click here to view a larger version of this figure.
Discussion
The clinical success of Imatinib in treating CML demonstrated not only the potential of targeting the rouge kinases by small molecule inhibitors, but also uncovered limitations of targeted therapy: clinical relapse and emergence of drug resistance mutations in the target gene. Identification of resistance mutations helps in better clinical management and development of next-generation inhibitors. This protocol describes a methodology to identify drug-resistant mutations in the targeted gene. This method uses a randomly mutagenized plasmid library built in E. coli, to express the mutant proteins. The library is then introduced in BAF3 cells (susceptible to transformation by an oncogene), followed with selection of cell clones in the presence of chemotherapeutic agents. Sequencing of targeted gene from resistant clones identifies mutations conferring resistance. Finally, identified mutations are recreated by site directed mutagenesis to validate them for drug resistance.
Using this strategy we defined the spectrum of mutations in JAK2 and BCR/ABL conferring resistance to Ruxolitinib and Imatinib, respectively. We identified more than hundred mutations in BCR/ABL. Besides, identifying all major mutations detected in patients, our screening method allowed us to identify novel substitutions of residues both within and beyond the kinase domain. Interestingly, all mutations from our screen have been identified in patient samples but this process took almost more than 10 years 15, thus demonstrating the ability of this screen to anticipate mutations that will pose clinically problematic drug resistance. While resistant screening using random mutagenesis provides many advantages in comparison to other methods, there are potential pitfalls to be aware of when following a screening procedure. For any screening, it is strongly recommended for BAF3 cells to be fully dependent on the target against which resistance is sought. A failure or partial dependence on the target gene may not develop true resistant clones and most likely develop false positives. For instance, four different studies have performed resistant screening against JAK2 inhibitors using BAF3 cells transduced with either EPO or MPL receptors to facilitate JAK2-V617F dependence 16-19. Only eight resistant mutations limited to kinase domain were identified from these screens, although numerous resistant clones developed that lacked mutations, presumably due to emergence of false positives, which was attributed to heterodimerization of JAK1 and JAK3 with cytokine receptors. Therefore, to avoid loss of JAK2 dependence we carried out resistant screening in plain BAF3 that showed robust selection of resistant clones against Ruxolitinib and all of these clones showed presence of resistant mutations. Based on these observations, in order to harness the full spectrum of resistant mutations, we recommend that the BaF3 cells should be fully dependent on the targeted kinase to avoid the emergence of false positives, as has been a norm in previous resistant screenings performed against JAK2 inhibitors16,17. Additionally, it is critical to avoid bulk culture conditions in which cells harboring different mutations are pooled together and allowed to expand in liquid culture, since this can lead to clonal dominance of a few highly drug resistant variants. For instance, during initial selection for imatinib-resistance of mutagenized BCR-ABL in bulk liquid culture, we found only 4 mutant forms that were represented multiple times within the first 100 isolates we sequenced. Therefore, screening using soft agar allows slow growing clones with more modest degrees of drug resistance to be recovered that would not be identified using bulk culture. For this reason, it is recommended to grow bulk culture prior to selection only for 14 to 16 hr, followed with drug selection without IL3 in soft agar. In our experience, growing the cultures beyond 36 hr after viral transduction tend to be dominated by highly proliferative clones, which poses a risk to lose slow growing clones (weakly transformative). Likewise, using cells from an earlier time points such as 6-8 hr after viral transduction are prone to select for highly transformative or overexpressing clones, thus causing a bias and misrepresentation in the identity and frequency of resistance clones. Therefore, we recommend using cells grown for 14-16 hr post viral transduction for screening, which provides tight selection of individual clones and prevents clonal dominance typically observed in bulk liquid cultures.
It is essential to confirm a candidate mutation’s ability to confer resistance in isolation, since some clones were identified to have multiple mutations. For the JAK2-V617F screen, we recreated a majority of the mutations identified from our random mutagenesis library using site directed mutagenesis. After introducing the isolated mutants into fresh cells, they were then grown in the presence of various drugs to determine IC50s for each mutant. We also tested two different resistant variants for in vivo resistance by monitoring the bioluminescence to further confirm the ability of these single mutations to cause drug resistance in vivo.
Given the success of anti-kinase therapy in treating cancers that galvanized a surge of kinase inhibitors in clinics, it is crucial to identify the clinically problematic resistance conferring mutations for better patient management and developing drugs that can target them. This screening strategy has been successfully used to identify mutations in the BCR/ABL, JAK2 and FLT3 20; however, we believe that it will be generally applicable to a broad range of anti-neoplastic agents.
Declarações
The authors have nothing to disclose.
Acknowledgements
This study was supported by grants to M.A. from NCI (1RO1CA155091), NHLBI (1R21HL114074) and the Leukemia Research Foundation. M.A. is a recipient of V-Scholar award from the V- Foundation. Authors are thankful to Dr. Sara Rohrabaugh for editing.
Materials
| name of Materials/Equipment | Company | Catalog Number | Comments/ Description |
| Cell and Tissue culture | |||
| BaF3 Cells | ATCC | ||
| HEK293T cells | ATCC | ||
| pMSCV-JAK2-V617F-puro.GW | A gift from Ross Levine | ||
| pMSCV-JAK2-V617F/Y931C.GW | Made in house | ||
| pMSCV-JAK2-V617F/L983F.GW | Made in house | ||
| pMSCV-JAK2-V617F/P58A.GW | Made in house | ||
| pMSCV-V617F-Cherry.GW | Made in house | ||
| pMSCV-JAK2-V617F/Y931C-cherry.GW | Made in house | ||
| pMSCV-JAK2-V617F/L983F-cherry.GW | Made in house | ||
| pMSCV-Luciferase-puro.GW | Made in house | ||
| RPMI | Cellgro (corning) | 15-040-CV | |
| DMEM | Cellgro (corning) | 15-013-CV | |
| Penn/Strep | Cellgro (corning) | 30-002-CI | |
| FBS | Atlanta biological | S11150 | |
| Trypsin EDTA 1X | Cellgro (corning) | 25-052-CI | |
| 1XPBS | Cellgro (corning) | 21-040-CV | |
| L-Glutamine | Cellgro (corning) | 25-005-CL | |
| Puromycin | Gibco (life technologies) | A11138-03 | |
| Protamine sulfate | Sigma | P3369 | 5mg/ml stock in water |
| Trapan Blue solution (0.4%) | Sigma | T8154 | |
| DMSO | Cellgro (corning) | 25-950-CQC | |
| INCB018424 (Ruxolitinib) | ChemieTeK | 941678-49-5 | |
| WST-1 | Roche | 11644807001 | |
| 0.45uM acro disc filter | PALL | 2016-10 | |
| 70um nylon cell stariner | Becton Dickinson | 352350 | |
| Bacterial Culture | |||
| XL-1 red E.Coli cells | Agilent Tech | 200129 | |
| SOC | New England Biolabs | B90920s | |
| Ampicillin | Sigma | A0166 | 100mg/ml stock solution |
| Bacto agar | Difco | 214050 | |
| Terrific broth | Becton Dickinson | 243820 | |
| Agarose | Genemate | E-3119-500 | |
| Kits | |||
| Dneasy Blood& tissue kit | Qiagen | 69506 | |
| Expand long template PCR system | Roche | 1168134001 | |
| Wizard Sv gel and PCR clean up system | Promega | A9282 | |
| Pure Yield plasmid mini prep system | Promega | A1222 | |
| PCR Cloning System with Gateway Technology with pDONR 221 & OmniMAX 2 Competent Cells | Invitrogen | 12535029 | |
| Gateway LR Clonase Enzyme mix | Invitrogen | 11791019 | |
| Mouse reagents | |||
| Vivo-Glo Luciferin in-vivo Grade | Promega | P1043 | |
| 1/2cc Lo-Dose u-100 insulin syringe 28 G1/2 | Becton Dickinson | 329461 | |
| Mortor pestle | Coor tek | 60316 and 60317 | |
| Isoflorane (Isothesia TM) | Butler Schien | 29405 | |
| Instruments | |||
| NAPCO series 8000 WJ CO2 incubator | Thermo scientific | ||
| Swing bucket rotor centrifuge 5810R | Eppendorf | ||
| TC-10 automated cell counter | Bio-RAD | This is not necessary, one can use standard hemocytomemetr for cell counting | |
Referências
- Huse, M., Kuriyan, J. The conformational plasticity of protein kinases. Cell. 109, 275-282 (2002).
- Melnikova, I., Golden, J. Targeting protein kinases. Nat Rev Drug Discov. 3, 993-994 (2004).
- Cohen, P. Protein kinases–the major drug targets of the twenty-first century. Nat Rev Drug Discov. 1, 309-315 (2002).
- Dancey, J., Sausville, E. A. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov. 2, 296-313 (2003).
- Noble, M. E., Endicott, J. A., Johnson, L. N. Protein kinase inhibitors: insights into drug design from structure. Science. 303, 1800-1805 (2004).
- Druker, B. J., et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 344, 1038-1042 (2001).
- Druker, B. J., et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 2, 561-566 (1996).
- Gorre, M. E., et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 293, 876-880 (2001).
- Shah, N. P., et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2, 117-125 (2002).
- Branford, S., et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 99, 3472-3475 (2002).
- Azam, M., Latek, R. R., Daley, G. Q. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 112, 831-843 (2003).
- Azam, M., et al. Activity of dual SRC-ABL inhibitors highlights the role of BCR/ABL kinase dynamics in drug resistance. Proc Natl Acad Sci U S A. 103, 9244-9249 (2006).
- Azam, M., et al. AP24163 inhibits the gatekeeper mutant of BCR-ABL and suppresses in vitro resistance. Chem Biol Drug Des. 75, 223-227 (2010).
- Azam, M., Seeliger, M. A., Gray, N. S., Kuriyan, J., Daley, G. Q. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 15, 1109-1118 (2008).
- Soverini, S., et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leukemia research. 38, 10-20 (2014).
- Deshpande, A., et al. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia. 26, 708-715 (2012).
- Koppikar, P., et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 489, 155-159 (2012).
- Marit, M. R., et al. Random mutagenesis reveals residues of JAK2 critical in evading inhibition by a tyrosine kinase inhibitor. PLoS One. 7, 43437 (2012).
- Weigert, O., et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. The Journal of experimental medicine. 209, 259-273 (2012).
- Kesarwani, M., Huber, E., Azam, M. Overcoming AC220 resistance of FLT3-ITD by SAR302503. Blood cancer journal. 3, 138 (2013).
