ETHICS STATEMENT: This protocol follows the animal care guidelines of the University of Valencia in compliance with European directive 2010/63/EU.
1. Generation of LV for In Vivo Marking Studies (see Figure 1a)
CAUTION: The procedure described herein is biosafety level 2, therefore perform all the following procedures in a biohazard hood. Ensure that research personnel are appropriately qualified and trained in all procedures. Wear personal protective equipment, including gown, double gloves and suitable eye protection. Finally, thoroughly decontaminate all tools and surfaces that could have been in contact with viruses according to approved facility disinfection practices (by wiping with 70% ethanol, 10% bleach and/or autoclaving).
- Production of LV in Human Embryonic Kidney 293T Cells
- Start this protocol by preparing pure DNA for transfection. Prepare and purify each plasmid by double CsCl gradient centrifugation or other commercially available column methods yielding endotoxin-free DNA. In this protocol we have used the transfer vector plasmid pRRL-SIN-PPT.PGK.EGFP.Wpre. Recommended core packaging plasmids are pMDLg/pRRE and pRSV.REV and envelope plasmid pMD2G13,18,19.
- Twenty-four hr before transfection, plate 5 x 106 293T cells in Iscove's Modified Dulbecco's Medium (IMDM) (see Table of Materials) in a 10 cm plastic dish in order to obtain an approximately 1/4 to 1/3 confluent culture for transfection. Incubate at 37 °C in a humidified incubator in an atmosphere of 5-7% CO2.
- Replace the medium with fresh medium 2 hr before transfection.
- In a sterile 1.5 ml microcentrifuge tube mix 10 µg of transfer vector plasmid (containing the cDNA of the transgene or the shRNA to be delivered) with 2.5 µg of the pRSV.REV and 5 µg of the pMDLg/pRRE packaging plasmids, and 3.5 µg of the envelope plasmid pMD2G. Make up the plasmid solution to a final volume of 450 µl with 0.1x TE buffer (see Table of Materials) /dH20 (2:1). Then add 50 µl of 2.5 M CaCl2.
- Form the precipitate by dropwise addition of 500 µl of the 2x Hepes Buffered Saline(HBS, see Table of Materials) solution to the 500 µl DNA-TE-CaCl2 mixture while vortexing at full speed.
- Add the precipitate to the 293T cells immediately. Gently swirl the plate to mix. Return the cells to the incubator and change the medium 14-16 hr after transfection.
- Collect the cell supernatants 30 hr after changing the media. Filter supernatant through a 0.22 µm pore nitrocellulose filter and proceed to concentration.
- Concentration of LVs
- Concentrate the conditioned medium by ultracentrifugation at 50,000 x g (19,000 rpm with SW-28 ultracentrifuge rotor) for 2 hr at room temperature (RT) in a 30 ml polypropylene transparent conical rotor tube.
Note: Use ultracentrifuge adapters for conical rotor tubes (see table of Materials). - Discard the supernatants by decanting and resuspend the pellets in a small volume (200 µl or less if only one centrifugation is performed) of phosphate buffer saline (PBS; see Table of Materials). Then pipette up and down about 20 times.
- Pool the suspensions and concentrate again by ultracentrifugation, also at 50,000 x g (23,000 rpm with SW-55 ultracentrifuge rotor) for 2 hr at room temperature. Use polypropylene transparent rotor tubes with a nominal volume of 5 ml (see Table of Materials).
- Resuspend the final pellet in a very small volume (1/500 or 1/1,000 of the starting volume of medium) of sterile PBS and shake on a rotating wheel for 1 hr at RT. Split into small aliquots (5-20 µl) and freeze them at -80 °C.
- Treat all empty tubes with 10% bleach before discarding.
- Concentrate the conditioned medium by ultracentrifugation at 50,000 x g (19,000 rpm with SW-28 ultracentrifuge rotor) for 2 hr at room temperature (RT) in a 30 ml polypropylene transparent conical rotor tube.
- Lentiviral Titration Using Flow Cytometry
- The day before, plate 5 x 104 HeLa cells per well in 6-well tissue culture plates in 2 ml Dulbecco's Modified Eagle's Medium (DMEM) (see Table of Materials). Incubate at 37 °C in a humidified incubator in an atmosphere of 5-7% CO2 for 24 hr.
- On the day of titration, thaw an aliquot of the viral stock and prepare serial dilutions, from 10-3 to 10-8, in DMEM.
- To do so, take a 24-well plate and add 2 ml of DMEM to the first well and 1.8 ml to the following wells. Then add to the first well 2 µl of the concentrated viral stock (to a final dilution of 1:1,000 or 10-3).
- After pipetting several times to thoroughly mix the solution, change tip and transfer 200 µl of the 10-3 dilution to the second well. Repeat the procedure serially in the following wells until the 10-8 dilution is made.
- Take HeLa cells plated the previous day from the incubator. Carefully remove medium from wells. Add 1 ml of each viral dilution together with 1 µl of 8 mg/ml hexadimethrine bromide to the HeLa cell-containing wells. Gently swirl the plate to mix.
Note: Hexadimethrine bromide is added to increase the virus adsorption to the cells in culture. - Return the cells to the incubator and allow the infection to proceed for 72 hr. After that, remove the medium, wash the cells once with PBS and add 200 µl of trypsin-EDTA (see Table of Materials) to each well.
- After 5 min at 37 °C, add 2 ml of PBS to each well and harvest cells in flow cytometry tubes.
- Centrifuge at 300 x g for 5 min at RT and aspirate the supernatant.
- Resuspend the pellet with 1 ml of fixing solution (1% formaldehyde electron microscopy grade and 2% fetal bovine serum in PBS), then vortex the tubes.
- Analyze the cells in a flow cytometer using a 488 nm argon-ionlaser at 15 mW power.
- Set up the instrument with the standard configuration: forward-scatter (FS), side-scatter (SS), and fluorescence for GFP (525/40 nm). Select cell population gating in a FS vs. SS dot plot to exclude cell aggregates and debris. Collect fluorescence in logarithmic scale. Calculate the number of GFP+ cells in each sample.
- Calculate vector titer using the following formula: % GFP+/100 x number of cells infected x dilution factor (DF) = transducing units (TU)/ml.
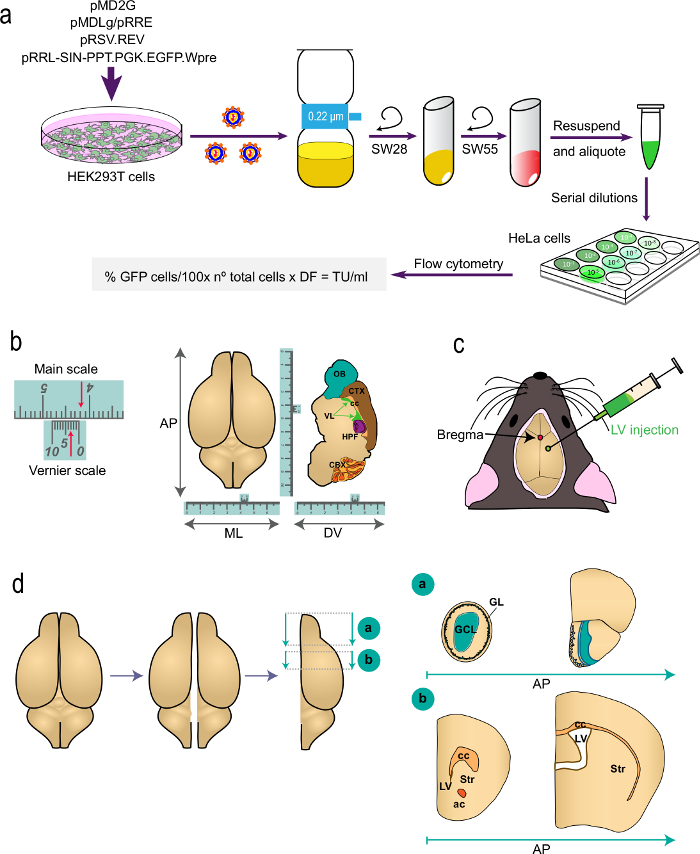
Figure 1: Schematic representation of the different parts of the procedure. (a) Part 1 of the protocol: generation of LVs for in vivo labeling studies, from the transfection of HEK293T cells with appropriate plasmids to generate the LVs to the determination of the virus titer by flow cytometry using the indicated formula. The names of the plasmids and the centrifuge rotors are indicated. (b and c) Part 2 of the protocol: stereotaxic injection of LVs. "b" depicts an example of a Vernier scale, a device that is part of stereotaxic instruments and serves for fine measurements. As an example, the arrows indicate 4.23 cm. A Vernier scale is used to determine the coordinates in the antero-posterior (AP), medio-lateral (ML), and dorso-ventral (DV) axis as shown for a top-view (left) and for a sagittal section (right) of the brain. "c" indicates the position of bregma as the intersection between the sagittal and coronal sutures. LVs are injected using a syringe. (d) Schematic drawings showing how the brain is processed for analysis. The two hemispheres are split and each one is divided into two blocks. Block "a", containing the OBs, is produced by a coronal cut at the AP level immediately posterior to the OB junction with the telencephalon (bregma 2.46 mm; see Paxinos´ Atlas for a reference). Block "b" is produced by two coronal cuts, one at the level just anterior to the most rostral aspect of the corpus callosum (bregma 1.7 mm) and a second one at the level of the junction of the two lateral ventricles (bregma -0.22 mm). GL, glomerular layer; GCL, granule cell layer; st, striatum; cc, corpus callosum; ac, anterior commissure; lv, lateral ventricle.
2. Stereotaxic Injection of LV into the V-SVZ/Striatum Border or into the Lateral Ventricle (see Figure 1b)
- Preparation
- Sterilize a 5 µl capacity syringe with a 33 gauge needle by spraying down the body and needle with 70% ethanol with the plunger pulled out all the way. Repeatedly aspirate ethanol from a 1.5 ml microcentrifuge tube and eject it all the way out several times, and rinse the syringe thoroughly with sterile water afterwards. Place the syringe safely aside in the culture hood and allow it to dry.
- Prepare a biohazard waste container with 10% bleach to a suitable volume for immersion of all waste from this procedure (generally 200 ml in a 500 ml container).
- Prepare and preheat a 37 °C waterbed by filling a sealable plastic storage bag with water and warming it to 37 °C. This will allow mice to recover following injection.
- Remove viral stocks from -80 °C freezer storage 1 hr before starting the injections and place the vial on a rotating wheel at RT. After thawing, maintain the viral stock on ice during the time of the injections. Prior to the stereotaxic injection of LV, dilute the concentrated viral stocks to 106 TU/μl using PBS in the culture hood.
- Sanitize the area selected for performing the surgery with 70% ethanol.
- Microinjection of LV
- Select and sterilize tools needed for surgery (scalpel, drill, and small tweezers).
- Anesthetize a 6-8 week-old mouse by intraperitoneally (ip) injecting a veterinary-supervised mixture of ketamine and medetomidine. Weigh each animal and dose each with 50-75 mg ketamine and 0.5-1 mg medetomidine per kg of mouse body weight (around 100-125 µl of the ketamine/medetomidine working solution per mouse).
- Assess the anesthetic plane by pinching the toes, tail or ear and ensuring that the animal shows no reaction.
- Once the mouse is anesthetized, inject butorphanol subcutaneously at a final dose of 0.4-0.5 mg per kg mouse weight to minimize post-surgical pain.
- Shave the area between the ears and disinfect the skin using an iodophor such as iodopovidone or 70% ethanol. Cleanse using sterile cotton-tipped applicators. Be careful not to excessively wet the animal as this can exacerbate hypothermia.
- Place the animal in prone position on a stereotaxic frame and carefully fix the head using the ear bars and the palate support of the apparatus. Keep the mouse with a heating pad set at 37 °C and apply ophthalmic lubricant to the eyes.
- Make a 1 cm long incision on the head skin longitudinally using a scalpel, and gently retract the skin to expose the skull using fine tweezers.
- Carefully clean the bone surface with a sterile cotton-tipped applicator. Cleanse the exposed skull bone of any remaining tissue.
- Mount the sterilized syringe on the stereotactic device using the syringe holder.
- Move the syringe holder x, y and z axis until the tip of the syringe needle is positioned on the bregma, the conjunction point where the sagittal (longitudinal and medial) suture is perpendicularly intersected by the coronal suture (Figure 1b). Ensure that the "zero" position of the dorso-ventral (DV) axis is at the skull surface at bregma.
- Move the syringe to the x and y destination coordinates (see Table 1 and Figure 1b).
| Region of injection | Coordinates | ||
| Antero-posterior (AP) | Medio-lateral (ML) | Dorso-ventral (DV) | |
| SEZ/striatum border | +0.6 mm | +1.2 mm | -3.0 mm |
| Lateral ventricle | -0.3 mm | +1.0 mm | -2.6 mm |
Table 1: Stereotaxic coordinates for the injections. For the AP and ML axis, x and y coordinates are given as a distance (in mm) from bregma. "-" indicates "towards posterior". For the DV coordinates "zero" is the surface of the skull at the bregma point and DV coordinates indicate the distance (in mm) down from this point.
- Annotate the x, y and z destination coordinates in the Vernier scale in order to be able to come back to the injection site later on. Mark the bone at the x and y coordinates using a surgical marker pen.
- Move the syringe away from the working area.
- Using an electric drill make a hole on the skull carefully not to damage the brain. Do not drill the pial surface as this may damage the brain surface.
- Load the syringe with 1 µl of the 106 TU/µl viral solution. Use a 33 gauge sharp beveled needle whose tip has an angle of 10-12°. Position the syringe needle at a 90° angle with respect to the brain surface.
- Move the syringe back to the site of injection and move it down until the tip touches the pial surface.
- Penetrate the brain with the syringe to the z coordinate in the DV axis.
- Slowly release the viral suspension, at a rate of 0.2 µl/min, in order to minimize damage to the brain tissue due to excessive fluid pressure.
- Wait for 5-10 min to minimize the backflow of viral suspension and then retract the syringe very slowly. Blot any excess of liquid that may appear at the surface as a result of the retraction of the syringe using a laboratory wipe and place it immediately in the bleach-containing biosafety waste container.
- Take the animal out of the stereotaxic set, place it on a warm pad, and close the wound using skin adhesive. Reverse the sedation using 0.1-1.0 mg/kg body weight atipamezole.
- Inject Bupenorphrine subcutaneously at a final dose of 0.1 mg per kg mouse weight every 12 hr, starting 4 hr after the administration of the short lasting Butorphanol analgesic.
- Place the animal in an individualized cage with a warm pad and monitor closely until the mouse recovers from anesthesia. Place one bag of hydrogel in the cage to help the animal hydrate after recovery.
- Dispose of all bio-contaminated waste in the liquid bleach biohazard disposal. Clean the syringe by aspiration and ejection of ethanol and rinse with water. Disinfect the area, the stereotaxic set and the surgical material that has been used with bleach and 70% ethanol.
- Keep injected mice isolated in the biosafety level 2 room for 24-48 hr after which they can be transferred to a conventional housing facility
3. Histological Analysis
- Perfusion, tissue collection, and sectioning
- Deeply anaesthetize the mice using a veterinary-supervised mixture of medetomidine and ketamine (assess the anesthetic plane by pinching the toes, tail or ear), as described before.
- Transcardially perfuse the mice with 25 ml of saline solution followed by 75 ml of 4% PFA in PB at the same rate17.
- Extract the brain and post-fix it by immersing it in at least 10 times its volume of cold 4% PFA in PB for 1-16 hr (increased post-fixation times may decrease the immunoreactivity of some antigens). Wash thoroughly the remaining PFA with PB.
- Cut the brain following indications of Figure 1d and glue the resulting block to the holder of a vibratome using cyanoacrylate.
- Collect 30 µm-thick serial coronal sections using a vibratome. Store the brain slices in 24-multiwell plates with PB at 4 °C. To prevent contamination, 0.05% sodium azide can be added to the PB solution.
- Immunohistochemistry
- Incubate the free-floating sections in blocking buffer (PB with 0.05% sodium azide, 1% glycine, 5% normal goat serum, and 0.1% Triton X-100) for 1h at RT with gentle shaking in a rocking platform.
- Carefully remove the blocking buffer with a pipette, add an appropriate dilution of anti-GFP rabbit primary antibody (see Table of Materials) in blocking buffer and incubate tissue with this dilution for 48 hr at 4 °C with gentle shaking.
- Wash off the primary antibody solution a minimum of 3 times with PB, one wash every 10 min.
- Incubate the free-floating sections with a suitable dilution of fluorophore-conjugated secondary antibodies) in blocking solution (see Table of Materials) for 1 hr at RT and gentle shaking. Protect the sections from direct light during the incubation.
- Wash off the secondary antibody solution with PB, 3 times once every 10 min, and counterstain the tissue by incubating the sections with DAPI (4',6-diamidino-2-phenylindole) at 1 mg/ml in water for 5 min. Wash off the DAPI solution by rinsing twice and quickly with water.
- Gently place the sections on a microscope slide using a fine paint brush. Pour a few drops of mounting medium for fluorescent preparations (see Table of Materials) over the tissue and carefully place a coverslip on top, checking that the mounting solution is correctly distributed over the entire surface and there are no bubbles. Gently squeeze down the coverslip to drain the excess of mounting medium.
- When the mounting solution dries out (2-16 hr), analyze the sample by confocal laser scanning microscopy with the 488 nm laser.
LV-mediated gene delivery system can be used for the long-term in vivo transduction of cells in the adult mouse V-SVZ, allowing their tracking and genetic modification during proliferation, migration and differentiation. The infection and the expression are highly effective and yield numerous cells that can be easily distinguished among other non-infected cells by the expression of the reporter included. We have thus far visualized transduced cells with GFP fluorescent reporters, driven by the ubiquitously expressed phosphoglycerate kinase promoter, but other reporters or protein tags can also be used. We routinely use antibodies to GFP instead of relying on the direct detection of the reporter emission as it yields a stronger fluorescent signal. Moreover, the brains can be dissected fresh and immersed in fixative but much better results are obtained by transcardial perfusion.
Purified, concentrated vector stocks are slowly injected under stereotaxic guidance into the V-SVZ at its limit with the striatum (Figures 2a, b). Two months after injection, numerous GFP+ cells were found in the V-SVZ and in an approximate 1 mm radius from the injection site (Figures 2a, b). Infection with LVs results in the transduction of all types of cells in the V-SVZ and expression of the reporter by ependymal cells indicates that LVs can also efficiently target non-dividing cells (Figure 2c; see also 15). GFP-labeling pattern in the V-SVZ was similar to that found after 2 weeks18. No sign of tissue reaction, however, was detectable at 2 months post injection since the long period allows the recovery of the tissue from the injury inflicted by the surgery. During the two-month period, infected TAP cells progress into A-neuroblasts that migrate out the zone towards the OB. Because this process takes only a few days3, the presence of GFP+ neuroblasts in the RMS 60 days after the infection indicates that LVs also target neurogenic B1-NSCs (Figure 2d). The transduction of B1, C, and A cells at the time of injection yields GFP+ interneurons in the OB (Figure 2e).
Injection of a small volume of high-titer concentrated LV stocks into the lateral ventricle results in the infection of ependymocytes only (Figures 2f-h). Most ependymal cells, which contain the calcium binding protein S100β, express the reporter after only 15 days from infection (Figure 2i). In contrast to the injection into the V-SVZ/striatal limit, no GFP+ cells were observed subependymally located or in the RMS or OB using this procedure (Figures 2h, j). Both types of infections can be analyzed in sections through the V-SVZ as explained. They can also be analyzed in whole-mount preparations of the lateral ventricle wall (see protocol in 8).
Recently, we have described how N-cadherin mediated cell-cell adhesion of B1 cells to ependymocytes regulates their quiescence. We have used the procedure described here to deliver LVs carrying cadherin dominant-negative constructs that resulted in inactivation of N-cadherin. We have demonstrated that loss of N-cadherin in the NSCs and ependymal cells or in ependymal cells only promoted an increase in the number and cell cycle entry of NSCs as a result of the disruption of homotypic cell-cell interactions (Figures 3a, b; see 15).
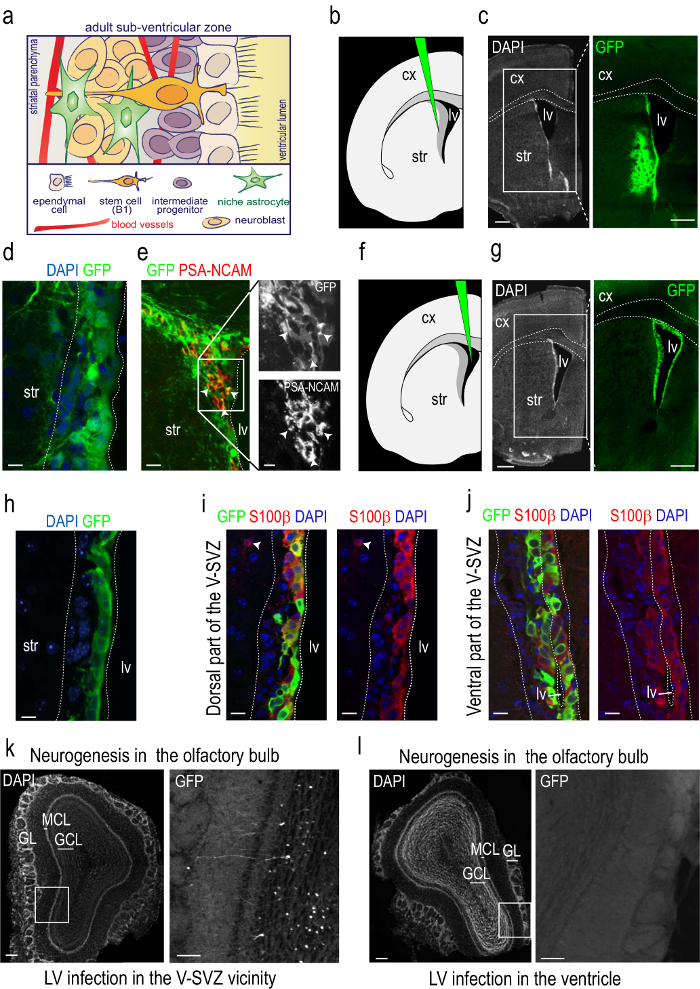
Figure 2: Reporter detection by immunofluorescence in sections of injected mice. (a) Schematic representation of the adult sub-ventricular zone niche. A simplification of the cellular components of the niche is depicted. B1 cells are polarized and exhibit a specialized relationship with the niche. They span between the ependyma which lines up the ventricle and the network of blood vessels that irrigate the V-SVZ niche. The small apical process of B1 cells intercalates among multiciliated ependymocytes and ends in a single non-motile primary cilium, whereas their basal process extends to approach the planar vascular plexus that irrigates this niche ending in the basal lamina of the plexus capillaries. B1 cell derivatives are intermediate progenitors that actively proliferate in the surroundings of blood vessels and migrating chains of neuroblasts towards the olfactory bulbs in contact with niche astrocytes. (b) Schematic of a coronal section through one hemisphere showing the level of injection and the position of the syringe needle for injections at the V-SVZ/striatum border. (c) Micrograph taken at the same level shown in the schematic stained with DAPI (gray; left) and for the immunodetection of GFP (green; right) 60 days after the injection. The dotted lines indicate the limits of the corpus callosum. (d) Higher magnification of an immunofluorescent detection of the GFP reporter injected in the V-SVZ vicinity or striatum border in a coronal section through the V-SVZ. The dotted lines indicate the limits of the V-SVZ. Note that many cells are infected in the V-SVZ niche. Ependymal cells can be recognized as cuboidal cells limiting the ventricle lumen. (e) Immunofluorescence for GFP (green) and the neuroblast marker PSZ-NCAM (red). Note the presence of doubly positive cells, indicating the generation of neuroblasts from NSCs that were infected 60 days before. White arrowheads point at neuroblasts. Insets correspond to the region enclosed within the white square. (f) Schematic of a coronal section through one hemisphere showing the level of injection and the position of the syringe needle for injections at the lateral ventricle. (g) Micrograph taken at the same level shown in the schematic stained with DAPI (in grey; left) and for the immunodetection of GFP (in green; right) 15 days after the injection. The dotted lines indicate the limits of the corpus callosum. (h) Higher magnification of an immunofluorescent detection of the GFP reporter injected in the ventricle in a coronal section through the V-SVZ. The dotted lines indicate the limits of the V-SVZ. Note that only ependymal cells are infected. (i–j) Immunofluorescence for GFP and the ependymal cell marker S100β. Note the presence of numerous doubly positive cells along the dorsal (i) and ventral (j) V-SVZ. Arrowhead points at an astrocyte in the striatal parenchyma, also labeled with S100β. k. Coronal section through the OB stained with DAPI (gray; left) and immunodetection of GFP (green; right) in the region enclosed by the white square 60 days after a V-SVZ/striatum injection. Notice numerous labeled neurons. (l) Coronal section through the OB stained with DAPI (gray; left) and immunodetection of GFP (in green; right) region enclosed by the white square 45 days after a lateral ventricle injection. Notice that there are no OB neurons labeled. cx, cerebral cortex; lv, lateral ventricle; str, striatum; GL, glomerular layer; GCL, granule cell layer; MCL, mitral cell layer. Scale bars: c, g = 500 µm; d, e, h-j = 10 µm; k-l = 250 µm; 100 µm.
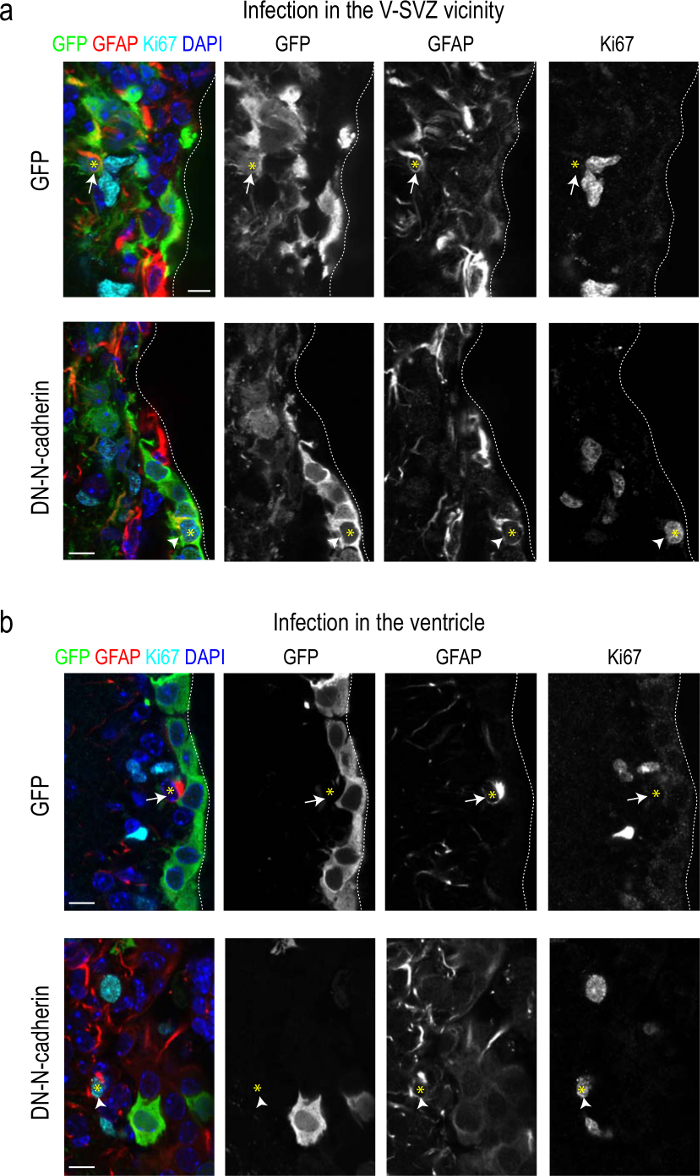
Figure 3: Adult NSCs proliferate more actively when they become detached from their niche. (a) Confocal micrographs of sections through the V-SVZ of mice that received striatal injections of empty LVs (upper panel) or LVs carrying a dominant-negative cadherin construct (bottom panel), immunostained for the detection of the LV reporter GFP (green), the NSC marker GFAP (red), the cell cycle marker Ki67 (cyan), and DAPI (blue) 60 days after the infections. Independent panels for each marker are shown (grey; right). (b) Confocal micrographs of sections through the V-SVZ of mice that received injections of empty LVs (upper panel) or LVs carrying a dominant-negative cadherin construct (bottom panel) into the lateral ventricle immunostained for the detection of the LV reporter GFP (green), the NSC marker GFAP (red), the cell cycle marker Ki67 (cyan), and DAPI (blue), 15 days after the infections. Independent panels for each marker are shown (gray; right). White arrows point at GFAP/GFP double positive cells and white arrowheads point at GFAP/Ki67/GFP triple positive cells whose nuclei in both cases are indicated by a yellow asterisk. Quantitative analysis indicated more cycling NSCs when the N-cadherin levels were decreased in the NSCs themselves or only in ependymal cells15. Scale: 10 μm.
