Figure 1 depicts different key steps of assessing cells and solutions during the transfection process. Figure 1a shows the optimal confluency of HEK293T cells prior to transfection for virus production. It is important that the cells are distributed evenly throughout the dish. Figure 1b reveals the precipitate in a drop of the transfection mixture using bright field optics and a 10X objective. The more the precipitates look like sand grains rather than agglomerates, the more efficient the transfection will be. In this picture, the precipitate is a bit coarse. The precipitate is a reliable indicator whether the transfection will produce a high-titer of virus. If any of the ingredients is either missing or of poor quality (vector DNA for instance), agglomerates will form, which is an ominous sign. If this is the case, it is wiser to abort and restart the procedure, making sure that all the ingredients are in the mix, and/or checking the quality of the vector DNA and packaging DNA. Figure 1c shows the same dish as A, after overnight incubation. The precipitate is (and should be) visible in between cells. We noticed that the cells do not multiply much during the 24 hr following transfection.
Figure 2 shows how HBE cells look before and 3 days after transduction. Figure 2a depicts approximately 80% confluent primary HBE cells prior to trypsinization and transduction. A 10 cm dish at 80% confluence yields approximately 4 x 106 cells. Figure 2b shows nearly 100% transduction of undifferentiated P1 HBE cells using an MOI of 4 on permeable support insert. When HBE cells are plated on these permeable support inserts, the cells are relatively flat. On this picture, the height of the cells is only a few microns which explains why the 0.4 µm pores are visible though the cells.
Figure 3 shows optimization experiments leading to the presented protocol. All experiments have been performed in triplicates on collagen I coated wells (24-well plate), with 2 x 105 primary HBE cells per well. Most experiments used an MOI of 0.4 with a 2 µg/ml final concentration of hexadimethrine bromidein BEGM media. Fluorescent cells were counted 3 days after transduction. Even though the initial optimization of the protocol has been performed on plastic, the results have been confirmed on permeable support inserts. In addition, attempts to transduce fully differentiated P1 HBE cells did not succeed (data not shown), confirming previous reports. As seen in Figure 3a, a higher percentage of HBE cells are transduced in BEGM media compared to ALI. If the cells are plated the day before transduction in BEGM media, the efficiency is significantly decreased compared to transducing while cells are attaching (Figure 3b). When P1 HBE cells are plated the day before transduction in ALI media (instead of BEGM), the transduction efficiency decreases even further (data not shown). The difference between these 2 media is the concentration of calcium. BEGM media contains a low concentration of calcium (0.1 mM) compared to ALI media (1 mM). Calcium forms and maintains tight junctions and therefore, could make it difficult for the virus particles to cross 9 and access their receptors at the basolateral membrane. In fact, some infections were successful at low levels by disrupting tight junctions using ethylene glycol tetraacetic acid (EGTA) for instance 10. As stated previously, fully differentiated P1 HBE cells are refractory and can only be transduced with low efficiencies 5-7, which could be due to tight junctions in high calcium containing media. Another explanation for low transduction efficiency in fully differentiated cells is an active mucociliary transport mechanism where mucus could act as a barrier. Or, as reported by Bals et al., the stage of differentiation in which HBE cells are, is a determining factor of transduction efficiency 10. The fact that more cells get transduced in BEGM than in ALI media supports this statement (Figure 3a).
As seen in Figure 3b, an increase of almost 50% transduction efficiency was observed when the cells were transduced while attaching compared to cells plated the day prior. In their study, Ricks et al. showed an increase of 20% when transduction was performed after trypsinization 11. Together, these results suggest that media and the state of maturity of cells is key to transduction success.
The panel in Figure 3c, demonstrates hexadimethrine bromideplaying atrivial role in transduction efficiency. Nevertheless, its use is suggested in this protocol since it has no negative impact but could potentially be beneficial. Hexadimethrine bromide is a cationic polymer that is used in virology to enhance transduction efficiencies 12. Others have published the use of protamine, another cationic polymer, but no significant difference was found regarding transduction efficiency compared to hexadimethrine bromide 13. Hexadimethrine bromide does not impact growth nor differentiation 14-16.
Finally, different MOIs were investigated in Figure 3d: the highest transduction efficiency occurred with an MOI of 4. There was no significant difference between MOI 0.4 and 4, however, MOI 4 transduced 100% of HBE cells whereas MOI 0.4 only transduced 75%. The MOI defines how many virus particles will have the potential to infect one cell. There are different methods to determine the MOI. One of them is to define the number of transduction units (TU). Using an ELISA, the amount of p24 antigen can be determined in pg/ml. Viral p24 in picograms can be converted in TU, which are necessary to calculate the MOI. The MOI is calculated by dividing the number of TU by the number of cells. There are 10-100 transduction units (TU) per picogram p24. All references in the text regarding MOI are based on calculations with 100 TU per picogram p24. (http://tronolab.epfl.ch/webdav/site/tronolab/shared/protocols/TUvsp24.html).
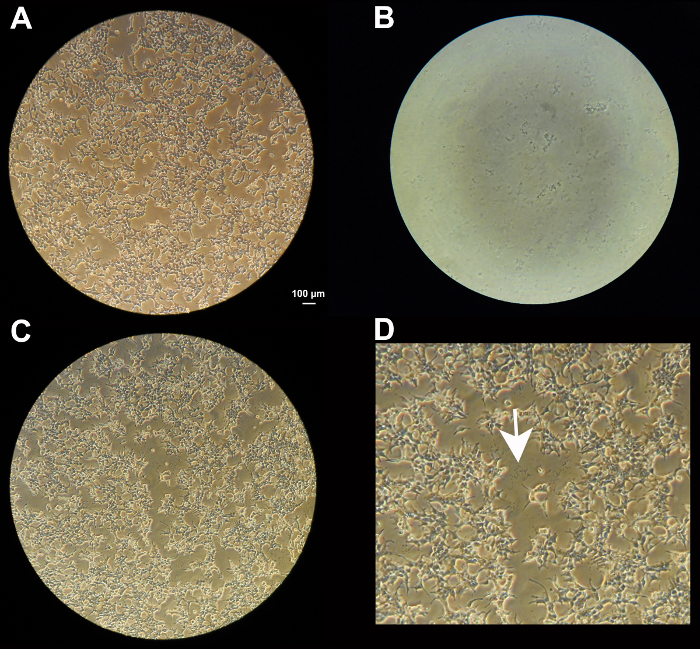
Figure 1: Transfection of HEK 293T cells. (a) HEK 293T cells grown to 50-60% confluency as seen under a 10X objective. (b) Precipitate visible in a 5 µl drop 5 min after mixing all reagents under the microscope (10X objective). (c) Precipitate in the dish next to the cells,following overnight transfection (10X objective). (d) Magnification of center of (c). White arrow pointing to precipitate. Scale bar = 100 µm; is the same for a, b, and c. Please click here to view a larger version of this figure.
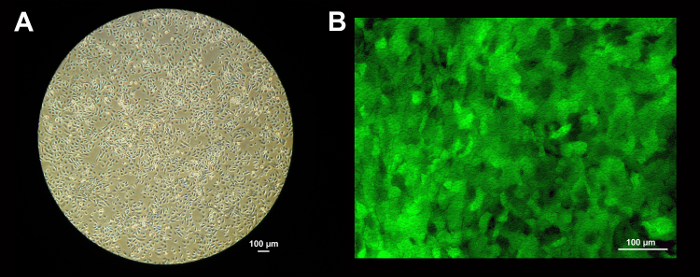
Figure 2: Transduction of NHBE cells. (a) HBE cells grown in a collagen I coated dish to 80-90% confluency (approximately 4 x 106 cells) in BEGM media for 5 days (10X objective). Scale bar = 100 µm. (b) eGFP transduced HBE cells on permeable support inserts (12 mm), 3 days post transduction (confocal, 20X). At this state, HBE cells are flat and the membrane's pores are visible. Scale bar = 100 µm. Please click here to view a larger version of this figure.
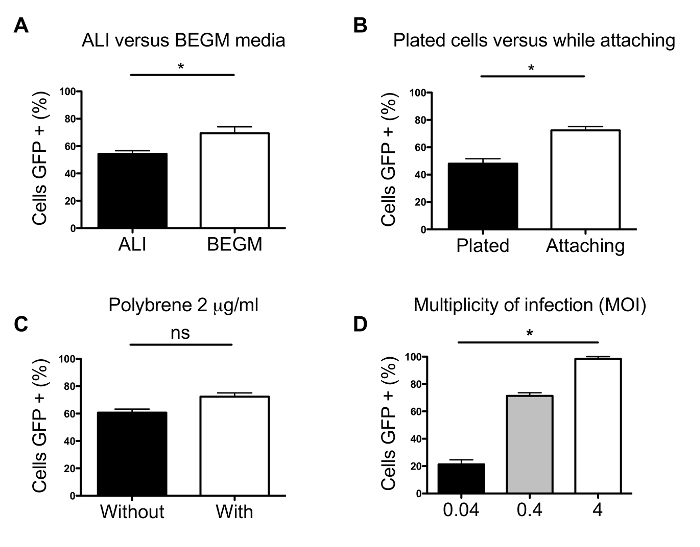
Figure 3: Optimization of transduction protocol. (a) Impact of media on transduction efficiency. (b) Comparison of transduction in HBE cells plated the day before and transduction of cells while attaching. (c) Influence of hexadimethrine bromide on transduction efficiency. (d) Evaluation of improved transduction efficiencies using different MOIs. Analysis was performed using Student's t-test or Kruskal-Wallis and Dunn's multiple comparison test. Error bars represent SE and all * represent p <0.05. Please click here to view a larger version of this figure.
