The protocol has to be carried out in strict accordance with national and international legislations and local regulations. Blood from healthy donors that gave their consent to donate blood for research purposes has been obtained from Blood Transfusion Centers with which the Institutions have signed agreement. Special protections must be taken when using human blood. Experiments with HIV-1 must be performed in a biosafety level 3 or 2 (BSL-3 or 2) laboratory according to local legislation.
1. Preparation of Human Monocyte-derived Macrophages (hMDMs) by Density Gradient Centrifugation and Selection by Adhesion
- Start with fresh blood from healthy donors (9 ml). Dilute the entire volume of fresh blood with sterile 1x phosphate buffered saline (PBS) without Ca2+ and Mg2+ to obtain a final volume of 70 ml and gently add the diluted blood into two 50 ml conical tubes (35 ml per tube), on top of 15 ml of a neutral, highly branched, high-mass, hydrophilic polysaccharide in solution already in each tube.
- Centrifuge both tubes of blood at 537 x g for 20 min at 20 °C without brake. Then collect the peripheral blood mononuclear cells (PBMCs) contained in the cloudy cell ring at the interface and transfer them into a new 50 ml tube containing 15 ml of 1x PBS without Ca2+ and Mg2+.
- Centrifuge the cells at 218 x g for 5 min at 20 °C and resuspend the pellet in 45 ml of 1x PBS without Ca2+ and Mg2+.
- Centrifuge the cells at 218 x g for 5 min at 20 °C, resuspend the pellet in 10 ml of 1x PBS without Ca2+ and Mg2+, and count the cells by diluting to a final dilution of 1/200 in Trypan Blue.
- Centrifuge the cells at 218 x g for 5 min at 20 °C and resuspend the pellet in RPMI (Roswell Park Memorial Institute) 1640 medium supplemented with 2 mM L-glutamine and 100 µg/ml penicillin-streptomycin to have 7 x 106 PBMCs per well in 2 ml of medium in each well of a 6 well-plate.
- Incubate the plates at 37 °C with 5% CO2 for 2 hr. After 2 hr the monocytes will have adhered to the plastic.
- To allow freshly-isolated monocytes to differentiate into hMDMs, aspirate the medium and replace it with 2 ml hMDM medium (RPMI 1640, 10% decomplemented Fetal Calf Serum (FCS), 2 mM L-glutamine and 1% penicillin-streptomycin) supplemented with recombinant human Macrophage Colony Stimulating Factor (rhM-CSF) at a final concentration of 10 ng/ml.
- Incubate the cells at 37 °C with 5% CO2 for 11 days (Figure 1).
- At day 11 remove the medium and wash 2 times with 2 ml of cold hMDM medium per well. Next wash each well 2 times with 1 ml of cold 1x PBS.
- To detach fully differentiated hMDMs, wash each well 1 time with 1 ml of cold 1x PBS with 2 mM ethylenediaminetetraacetatic acid (EDTA) and incubate the cells in 2 ml of cold 1x PBS with 2 mM EDTA per well for 15-60 min at 4 °C.
NOTE: Alternative methods to detach the cells exist (e.g., trypsin or trypsin-like activities) that might give good results 11. - After detachment (completed by pipetting gently up and down in the well), collect the cells and put them in a 50 ml tube on ice containing 10 ml of cold hMDM medium.
- Centrifuge the cells at 218 x g for 5 min at 20 °C, resuspend the pellet in 10 ml of cold hMDM medium, and count the cells by diluting 1/20 in Trypan Blue.
- Seed the cells at 1 x 106 hMDMs per 35 mm microscopy grade glass bottom dish and incubate the plates at 37 °C with 5% CO2 for 1 day.
2. Production and Quantification of HIV-1 Viral Stocks
NOTE: NLR4.3 HIV-1 Gag-iGFP (Green Fluorescent Protein) carrying an R5-tropic envelope, gift from M. Schindler 10 is used to infect macrophages and to see infected cells in real time.
- Produce viral stocks by transfection of human embryonic kidney 293T cells (2 x 106 in 100 mm dish) with 6 µg of the corresponding proviral DNA using a commercial transfection reagent.
- Quantify the infectivity of the virus stocks using the indicator cells HeLa TZM-bl (bearing the ß-galactosidase gene under the control of HIV-1 LTR) using serial dilutions of the viral stocks followed by a ß-galactosidase coloration of the cells and counting of blue cells 12.
3. Infection of hMDMs with HIV-1
- Add the virus at a multiplicity of infection (MOI) 0.02-0.03 in 1 ml of hMDM medium to hMDMs (cells plated in 1.13). To control wells add only 1 ml of hMDM medium and incubate the dishes at 37 °C with 5% CO2 for 2 days (Figure 1).
- At day 2 wash the cells with hMDM medium 3 times and add 1 ml of fresh hMDM medium per dish. Incubate the cells at 37 °C with 5% CO2 for 6 days (Figure 1).
4. Opsonization of Sheep Red Blood Cells
- For the preparation of 7 x 106 SRBCs per dish, wash the SRBCs two times in 100 µl of solution containing 0.1% Bovine Serum Albumin (BSA) in 1x PBS with centrifugation at 600 x g for 4 min.
- Resuspend the washed SRBCs in 500 µl 1x PBS/BSA 0.1% with rabbit IgG anti-SRBCs at a sub-agglutinating concentration per 5 µl of SRBCs and incubate with rotation at RT for 30 min.
NOTE: To determine the sub-agglutinating concentration of anti-SRBCs IgG, prepare serial dilutions of IgG (stock concentration at 13.1 mg/ml) from 1/50 to 1/25,600 in 20 µl in a 96-well plate. Add 2 x 106 SRBCs in 20 µl in each well and put the plate in a dark room during several hr. The sub-agglutinating concentration is the dilution of the well just before the well with agglutination (IgG+SRBCs forming a network). - After rotation, centrifuge the IgG-opsonized-SRBCs at 600 x g for 4 min and wash with 100 µl of 1x PBS/BSA 0.1% with centrifugation at 600 x g for 4 min.
- Resuspend the IgG-opsonized-SRBCs in pre-warmed Phenol red-free RPMI medium supplemented with 2 mM L-glutamine and 1% penicillin-streptomycin (1 ml/dish).
5. Live Cell Video Microscopy Phagocytosis Assay
- Use a confocal imaging system such as a spinning disk system equipped with a heating chamber at 37°C with CO2 passing through a bottle with water for humidification.
- Turn on the heating chamber prior to the experiment to have the microscope stage at 37 °C before the beginning of the phagocytosis assay. Turn on the microscope and computer, and load the imaging software.
- Optimize the imaging settings such as scanning speed, magnification, resolution, etc. to have at least one cell per field and to image one frame every minute between 60 to 120 min.
NOTE: Here, the sample is imaged one frame every minute during 60 min with 63X lens. - Place the imaging dish on the microscope stage. Adjust the focus and the location to find just one whole HIV-1 infected macrophage in the field. Use appropriate excitation/emission settings based on the used imaging system and probe. Include a bright field (BF) channel to observe phagosomes (Figure 1ii). Optimize the appearance of the different channel images by adjusting the percentage of transmitted light and exposure time.
NOTE: Here, NLR4.3 HIV-1 Gag-iGFP was excited using a 491 nm laser with 50 msec of time exposure with 20% of laser (Figure 1i). - Remove the imaging dish and add 1 ml of SRBC suspension at 7 x 106 SRBCs/ml to the dish (Figure 1).
- Centrifuge at 500 x g for 2 min at RT to synchronize phagocytosis, record the time at the end of this centrifugation and return the dish to the stage.
- Optimize the focus and capture GFP and BF images in Z-stacks (throughout the thickness of the cell with a step-distance of 0.3 µm – usually 20 planes) every minute for at least 1 hr. Save the time-lapse video in the native file-format of the used imaging system.
NOTE: Here, time-lapse movies were saved in the native format, *.stk files.
6. Analysis of the Time-lapse Movies
- For video editing, click on the drop down menu "Apps" and on the tab "Review Multi Dimensional Data" in the video editing software.
- To open the file, click on "Select Base File" then on "Select Directory". In the "Data sets" box, select the acquisition to analyze (in .nd format) and click on "View". The data are represented in a table with time in the column and Z-plan in the line.
- To represent the infection in a Z-projection (Figure 2, left panels), select 491 nm wavelength in the "Wavelengths" box and click on the "Z projection" tab with "all planes".
- To analyze a time sequence, select BF wavelength in the "Wavelengths" box and choose the optimal plane on the Z-axis to distinguish external SRBCs (Figure 2, red arrowhead), internal SRBCs (Figure 2, red circle) and the nucleus (Figure 2, blue circle).
- To save the video montages, click on "Selection [X's]" tab and then on "Load Image(s)". Finally, save the loaded images in .tif format and open them next on ImageJ software.
- Use the plugin "Manual Tracking" on the ImageJ software to measure the position of the nucleus and the different phagosomes observed in BF channel (Figure 3).
- Download the "Manual Tracking" plugin on the ImageJ website. On ImageJ, open the plugin and the image sequence to be analyzed (Figure 3, step 1 and 2).
- Enter the settings such as "Time Interval" which represents amount of time between adjacent frames, and the "x/y calibration" which represents the distance per pixel (Figure 3, step 3).
NOTE: Here, save the image sequence with a time interval of 1 min between each frame and x/y calibration of 0.205 µm because the 63X zoom and a camera with pixel size of 6.45 x 6.45 µm2 are used. - To start the tracking, click on "Add track" (Figure 3, step 4) and click on an SRBC center at the first time when it is internalized. The next frame appears automatically.
- Continue to click on the SRBC center in all frames to have different positions during the time (Figure 3, step 6 in red box).
- Start to track the nucleus to have its position in all frames by clicking on its center. Next track the phagosomes (by clicking on its center) one by one at the time (frame) of their internalization, different in function of SRBCs. For convenience to see SRBC on BF channel, use the "Brightness & contrast" window during the tracking (Figure 3, step 5).
- Between each SRBC tracking, click on "Add track" (Figure 3, step 4) to have a new track. The number of the tracking will be changed in the second column, "Track n°" of the results table (Figure 3, step 6).
- Save the data in a spreadsheet to continue to the next step of the analysis.
- Use spreadsheet software to calculate the travelled distance of phagosomes containing SRBCs towards the nucleus and the velocity of the phagosomes during the first 5 min after internalization of SRBCs (Figure 4).
- Open the spreadsheet table and a new spreadsheet file. Transfer into this new file the following parameters only, time, the x- and y- coordinate of the nucleus and all SRBCs (Figure 4A).
- To calculate the distance between SRBCs and the nucleus with only their coordinates, consider the nucleus and a SRBC in a XY-coordinate plane (Figure 4B).
NOTE: The distance between two points is the length of the path connecting them. In the plane, the distance between SRBC and the nucleus is given by the Pythagorean theorem.- If c (the distance between the nucleus and the SRBC) denotes the length of the hypotenuse and a and b denote the lengths of the other two sides, express Pythagoras' theorem as the Pythagorean equation:
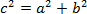
NOTE: At the same time, on the orthonormal, the horizontal distance a is (xnucleus-xSRBC) and the vertical distance b is (ynucleus-ySRBC). - Thus, calculate the distance between the nucleus and the SRBC (Figure 4A, purple box) by:
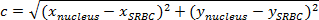
NOTE: Multiply the obtained distance value (in pixels) by x/y calibration to have the distance in µm. Here, the x/y calibration is 0.205 µm. - At each time, subtract the distance between the nucleus and the SRBC to the initial distance (Figure 4C).
- Plot the measurement against the time for the first 5 min. Apply a linear trend-line (Figure 5A) to the plot and determine the slope of the linear trend-line that represents the phagosome velocity towards the nucleus during the first 5 min after SRBC internalization.
- Collate the data to calculate the average and statistical error of the velocity values and plot them in an appropriate form using the appropriate software (Figure 5B).
- If c (the distance between the nucleus and the SRBC) denotes the length of the hypotenuse and a and b denote the lengths of the other two sides, express Pythagoras' theorem as the Pythagorean equation:
FcR-mediated phagocytosis by HIV-1-infected and non-infected hMDMs is described here using IgG-opsonized SRBCs as model targets (Figure 1). The critical steps of this protocol are the preparation of hMDMs and the infection with HIV-1. Indeed, the yield and quality of differentiated macrophages varies among donors, as well as the infection rate with efficiencies in the range of 10-40%. In addition, the preparation of IgG-opsonized-SRBCs is also important to avoid damaging the erythrocytes, because this could induce their recognition and uptake as debris instead of by FcR-mediated phagocytosis. Optimal opsonization will ensure efficient phagocytosis.
Phagosome movement in living HIV-infected macrophages is analyzed in real-time with the BF channel on a spinning disk confocal microscope in the BSL-3 laboratory. The settings of the BF channel are critical to see the nucleus (Figure 2, blue circle) and for discriminating the external (Figure 2, red arrowheads) from the internalized SRBCs (Figure 2, red circles). The BF microscopy is considered as the simplest optical microscopy illumination technique sufficient to see the phagosomes, which are less refringent than the external SRBCs.
The first step after acquisition of image sequences is manual tracking on the ImageJ software (Figure 3). This point can be particularly long and tedious, because it is preferable to track each phagosome, one by one for all image sequences and it is considered that experiments need to be repeated with at least 3 donors per condition to reach a large enough sample size for statistical analysis (at least 30 phagosomes with 3 different donors).
The second step is the analysis in spreadsheet software to calculate the phagosome velocity during the first 5 min after internalization for each internalized SRBCs (Figure 4). For this, we plot for each phagosome the distance travelled during the first 5 min against the time. A representative example is shown in non-infected hMDMs on Figure 5A. Next we obtain the velocity of the phagosome, which is the slope of the linear regression curve. A velocity of 0.5384 µm/min was found for this phagosome.
Of note, it is possible to analyze the phagosome velocity during the first few minutes after internalization, as described here or in Dumas et al. 8, or later by analyzing the velocity over longer timescales. In our study, we observed that the phagosome velocity during the first 5 min after internalization in HIV-infected hMDMs is lower compared to non-infected hMDMs (Figure 5B).
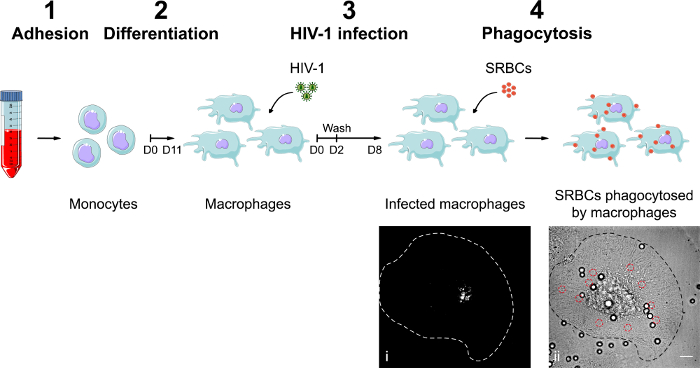
Figure 1. Preparation of control and HIV-infected hMDMs for phagocytosis imaging. Monocytes are purified by adhesion (1) and differentiated into macrophages for 11 days with M-CSF (2). Next, hMDMs are infected with NLR4.3 HIV-1Gag-iGFP i) at an MOI between 0.02 and 0.03 for 8 days with washing on day 2 (3). (4) IgG-opsonized SRBCs are added onto hMDMs for phagocytosis during 1 hr. The phagosomes containing SRBCs are detectable on BF images (red circle, ii). Images are from the Servier images database. Bar = 10 µm. Please click here to view a larger version of this figure.
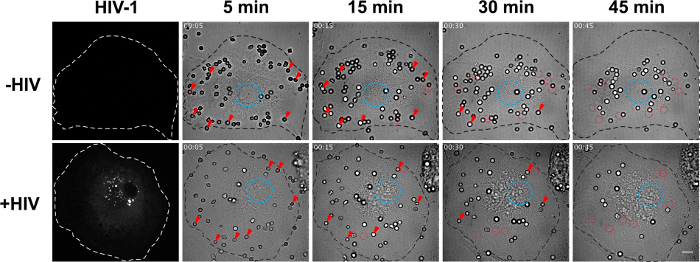
Figure 2. Real-time acquisition during phagocytosis by representative control and HIV-infected hMDMs. hMDMs are prepared and infected as described in Figure 1. The NLR4.3 HIV-1Gag-iGFP infected cells are detected with the 491 nm laser. Z-stacks of spinning disk confocal images are acquired and analyzed using both the microscope acquisition software and ImageJ (left panels). Galleries of BF images represent a non-infected (top panels corresponding to Movie 1) and a NLR4.3 HIV-1Gag-iGFP infected (bottom panels corresponding to Movie 2) cell during a 45 min acquisition (46 images with time indicated as hh:mm). The cell membrane (dotted black line), the nucleus (blue circle), the external SRBCs (some of them indicated with red arrowheads) and the internal SRBCs (some of them indicated with red circles) are detectable on BF images. Bar = 10 µm. Please click here to view a larger version of this figure.
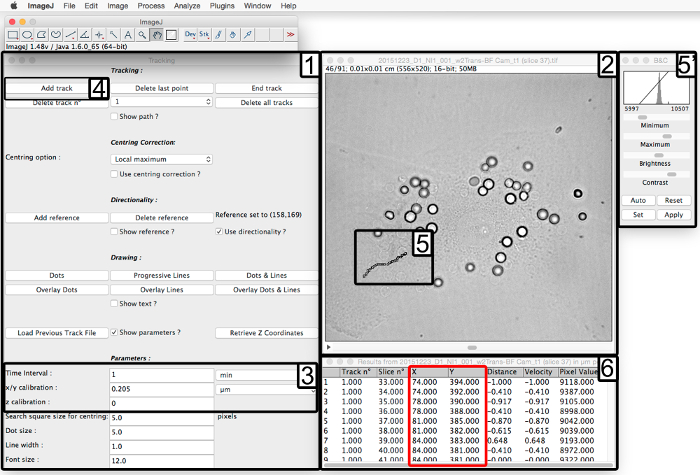
Figure 3. Instruction steps for image analysis with Manual Tracking plugin on ImageJ. After opening the "Manual Tracking" plugin (1) and loading the time-lapse video in BF channel (2), enter the parameters of the acquisition (3). Click on "Add track" (4) and start the measurement of the tracking by clicking on one phagosome (5) to obtain a new window with X and Y positions of the chosen phagosome (6, red box). To follow the phagosome on the time sequence for convenience, vary the brightness and contrast (5'). Please click here to view a larger version of this figure.
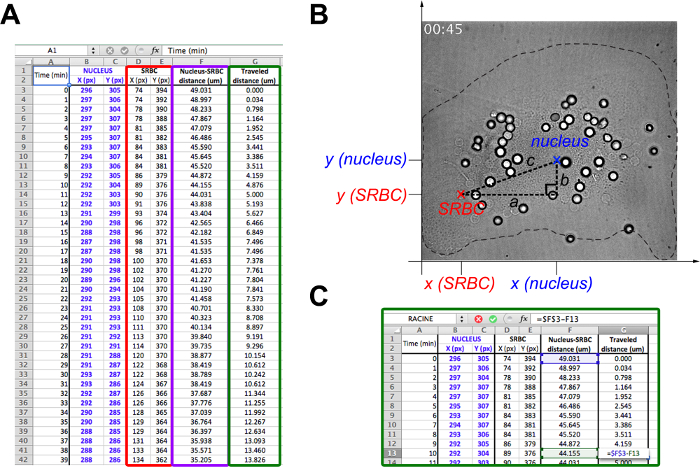
Figure 4. Instruction steps for data analysis of phagosome migration velocity in spreadsheet software. (A) Assemble the X and Y position of the nucleus and each phagosome (red box) in a spreadsheet table. In another column (purple box), calculate the distance between the phagosome and the nucleus with Pythagoras' theorem where c represents the length between the nucleus and the SRBC and a and b the lengths of the other two sides of a right-angled triangle (B). (C) Finally, calculate the distance travelled by the phagosomes at each time (green box) by subtracting the distance between the SRBC and the nucleus at a given time from the initial distance at time 0. Please click here to view a larger version of this figure.
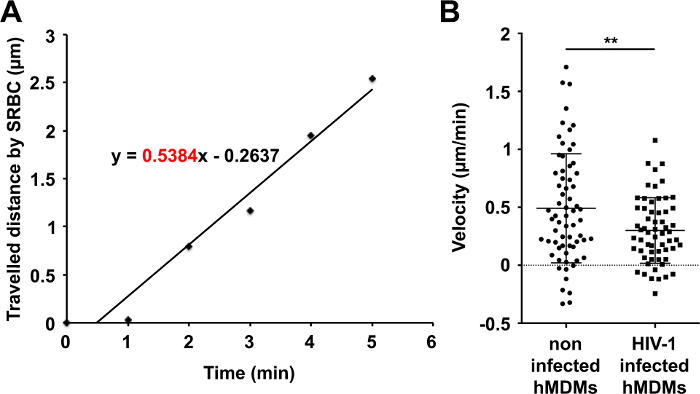
Figure 5. Quantification of phagosome migration velocity in control and HIV-infected hMDMs. (A) For each SRBC and just after SRBC internalization, the travelled distance towards the nucleus is measured and plotted against the time. Its velocity during the first 5 min is calculated using linear regression in spreadsheet software. A representative experiment is shown (the internal SRBC of Figure 3-4). (B) All velocities during the first 5 min are measured for 66 phagosomes in non-infected (5 donors) and 60 phagosomes in HIV-infected cells (4 donors). Data represent the mean ± SEM (Unpaired Student t test with Welch's correction; **, P <0,01). Please click here to view a larger version of this figure.
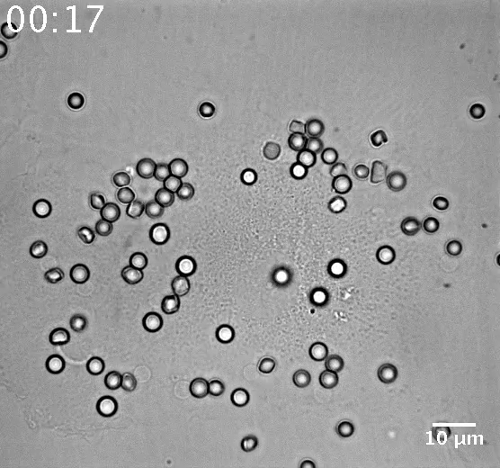
Movie 1: Phagosome movement in a representative non-infected macrophage. (Right click to download). The movement of opsonized red blood cells was detected in BF channel during 45 min of phagocytosis with one image per minute (46 images with time indicated as hh:mm). Bar = 10 µm.
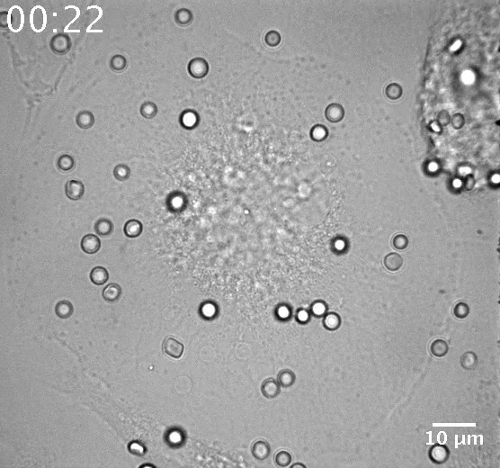
Movie 2: Phagosome movement in a representative HIV-infected macrophage. (Right click to download). HIV-1 (NLR4.3 HIV-1 Gag iGFP)-infected macrophages were identified after illumination with the 491 laser. The movement of opsonized red blood cells was detected in BF channel during 45 min of phagocytosis with one image per minute (46 images with time indicated as hh:mm). Bar = 10 µm.
