In this demonstration experiment, multimer-PAGE was performed on whole rat brain lysate. The resultant separated proteins were blotted onto polyvinylidene diflouride (PVDF) membranes, and then probed with antibodies against proteins that are known to form complexes. Figure 1 shows a validation of the protocol by two means. First, we demonstrate that the cross-linked proteins are cleavable by addition of a reducing agent, meaning the observed higher molecular weight species are formed by the cross-linking reagent, and are not due to any other component of the protocol (Figure 1A). Second, we demonstrate that cross-linking is sensitive to addition of detergents (Figure 1B), meaning the cross-linking is specific to complexed proteins and that random reactivity due to close proximity on the gel is minimized. Figure 2 demonstrates that the method fails to capture respiratory complex II, but otherwise has wide applicability for capturing both soluble and membrane-bound complexes.
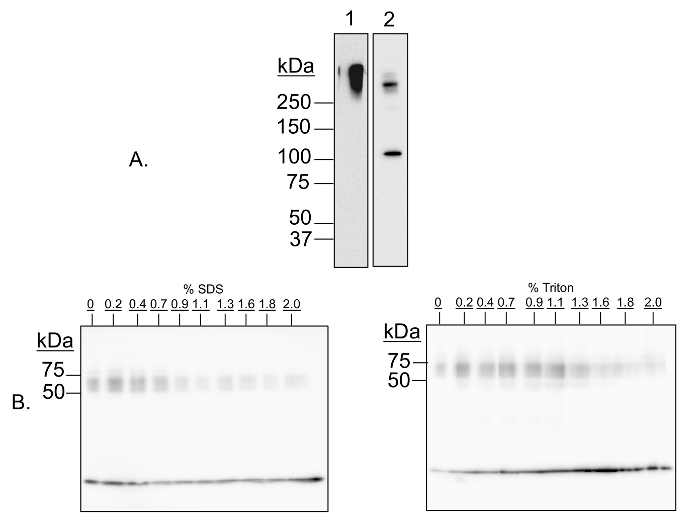
Figure 1: Validation of multimer-PAGE method. (A) Capture of protein complexes by in-gel cross-linking with DSP is sensitive to cleavage by dithiothretol (DTT). Multimer-PAGE was performed on whole rat brain lysate without (A1) or with (A2) 5 mM dithiothretol included in the Tris/SDS quenching solution. The gel was electroblotted onto PVDF membranes, and then probed for kinesin heavy chain. A high molecular weight band is observed on both membranes, likely corresponding to all or part of the kinesin motor protein complex. There is an additional band occurring at 126 kDa on the DTT-treated blot, the molecular mass of the kinesin heavy chain peptide. Dithiothreitol is a reducing agent, and is able to reverse cross-linking by cleaving the disulfide bond present in DSP. The kinesin aggregate is therefore sensitive to cleavage by DTT. (B) Protein complex capture is sensitive to addition of denaturing detergents. Multimer-PAGE was performed on whole rat brain lysate as described in the text, except increasing amounts (0 to 2%) of SDS (left) or Triton X-100 (right) were added to the lysate samples, and then incubated on ice for 30 min before loading the first gel. The final gels were blotted onto PVDF membranes, and probed for α-synuclein. At least three bands of increasing molecular weight are observed, corresponding to α-synuclein monomer, tetramer, and octamer, respectively. The signal intensities of the oligomeric bands decrease with increasing detergent concentration, indicating that cross-linking efficacy is dependent of native protein conformation. Please click here to view a larger version of this figure.

Figure 2: Demonstration of the capture of protein complexes using multimer-PAGE. (A) Soluble and membrane-bound protein complexes were captured from (A) whole rat brain lysate or (B) lysate incubated with 2% digitonin for 1 h at 4 °C, by performing multimer-PAGE as described in the text. The gels were then blotted and the PVDF membranes probed for components of various complexes. High molecular weight species are observed on the membrane probed for acetylated tubulin, which is known to stabilize microtubules. Dynein and kinesin are multi-subunit motor protein complexes with molecular weights of 1.5 MDa and 380 kDa, respectively. The membranes blotted for components of these complexes show high-molecular weight species. In addition, multimer-PAGE successfully captured the HSP90 dimer. α-Synuclein forms tetramers and octamers, as well as neurotoxic high-molecular weight oligomers. The octamer and a streak of higher-weight species are seen on the blotted membrane. VDAC dimerization is also observed. High molecular weight species are detected on the membranes blotted for components of mitochondrial complexes I and IV. However, the membrane blotted for SDHA, a member of complex II, does not demonstrate any appropriate high-molecular weight band. AcetTUB: acetylated α-tubulin; βTUB: β-tubulin; Dynein: dynein heavy chain; HSP90: heat shock protein 90; Kinesin: kinesin heavy chain; NDUFS3: iron-sulfur center 3 of mitochondrial complex I; VDAC: porin; SDHA: succinate dehydrogenase of mitochondrial complex II; COXIV: cytochrome C oxidase of mitochondrial complex IV. Please click here to view a larger version of this figure.
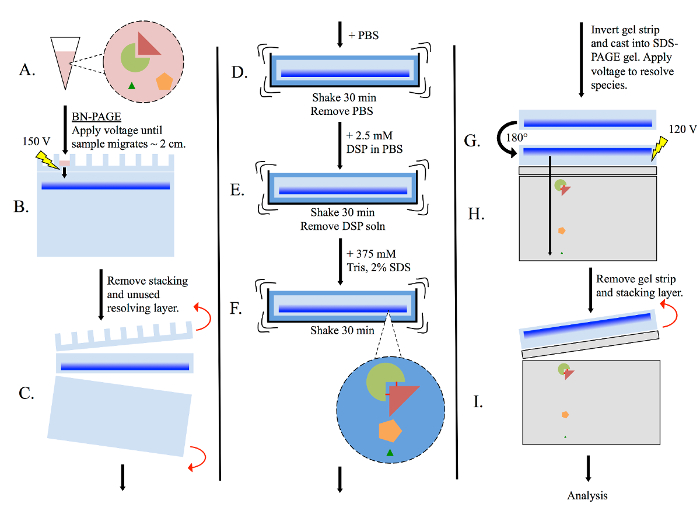
Figure 3: General multimer-PAGE flowchart. (A) Prepare tissue lysate using non-denaturing methods, such as homogenizing in BN-PAGE sample buffer. (B) Pipette lysate into BN-PAGE gel, then perform electrophoresis (150 volts) in BN-PAGE running buffer. Allow sample to migrate until the dye front has migrated ~2 cm into the resolving gel. (C) Remove the stacking layer and unused portion of the resolving layer. (D) Submerge the gel strip in PBS, and equilibrate with shaking for 30 min. (E) Remove the PBS, and re-submerge the gel strip in PBS containing 2.5 mM DSP. Shake for 30 min. (F) Remove DSP solution, then re-submerge the gel strip in 375 mM Tris containing 2% SDS, and shake for 30 min. Complexes present in the gel strip are now stabilized by cross-linking. (G) Rotate the gel strip 180° and cast into a new SDS-PAGE gel. Include both stacking and resolving layers. (H) Resolve sample by electrophoresis (120 volts). (I) Remove gel strip and stacking layer, then analyze resolving layer as necessary. Please click here to view a larger version of this figure.
