Using this platform, we investigated the role of both biochemical and biophysical cues in the fate specification of liver progenitors34,35. Protein A/G-conjugated Notch ligands showed improved retention and clustering in the polyacrylamide hydrogel (Figure 3A) and were furthermore capable of driving differentiation of liver progenitors towards a bile duct cell fate (Figure 3B). Using single-cell analysis, we quantified the response to the Notch ligands for the ECM proteins collagen I, collagen III, collagen IV, fibronectin, and laminin (Figure 3C), finding that the response of liver progenitors to the ligand depends also on the ECM context. Last, we utilized shRNA knockdown to generate liver progenitors without the ligands Dll1 and Jag1. The response to the arrayed Notch ligand varied depending on the presence of either ligand, confirming that the responsiveness to the cell-extrinsic ligand is also a function of the cell-intrinsic ligand expression (Figure 3D). Further, we observed a distinct subpopulation of double-positive (ALB+/OPN+) cells in the Dll1 knockdown (Figure 3D). Together, these representative results show: (1) the combinatorial capabilities of the array format, as exemplified by the pairing of multiple arrayed ECM proteins and Notch ligands with the knockdown of individual ligands; (2) the functionality of not only arrayed ECM proteins but also arrayed cell–cell ligand via Protein A/G-mediated conjugation; and (3) the implementation of our single-cell analysis and its ability to discern unique subpopulations.
We also observed that the differentiation of liver progenitors is dependent on both the substrate stiffness and the ECM composition (Figure 4A), specifically finding that collagen IV is supportive of differentiation on both soft and stiff substrates while fibronectin only supports differentiation on stiff substrates (Figure 4B). Representative heat maps of TFM measurements suggested that sustained traction stress at low substrate stiffness on collagen IV promoted differentiation into bile duct cells (Figure 4C), a finding confirmed by average root-mean-square values (Figure 4D). Together, these representative results show: (1) the successful integration of TFM with cell microarrays on substrates with a tunable stiffness to assess both the cell phenotype and the traction stress; (2) the coordination of the liver progenitor cell fate with both the matrix composition and the substrate stiffness; and (3) the implementation of our TFM analysis and typical traction stress profiles in cell microarrays.
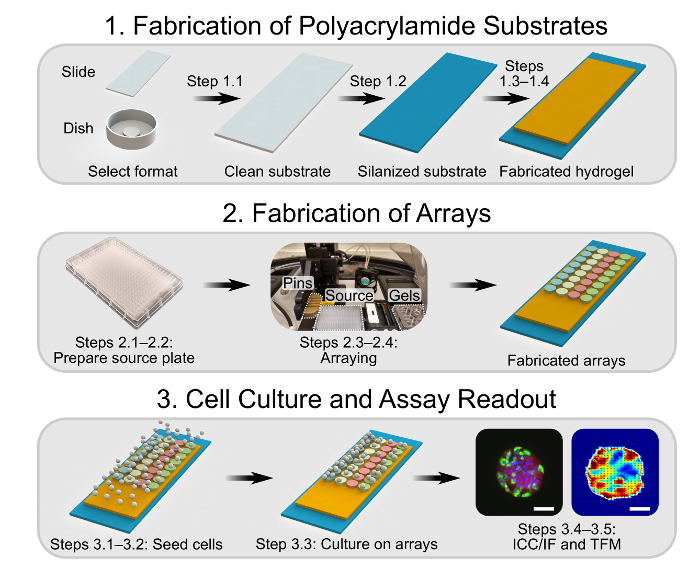
Figure 1: Overview Schematic Showing the First Three Experimental Sections. In Section 1, glass substrates are cleaned and silanized to facilitate the fabrication of polyacrylamide hydrogels. In Section 2, the biomolecule combinations of interest are prepared in a 384-well source microplate. A robotic arrayer is then loaded with clean pins, the source microplate, and the polyacrylamide hydrogels and initialized, fabricating arrays on the hydrogels. In Section 3, cells are seeded onto the arrayed domains and allowed to adhere, after which the culture protocol of interest is performed. At the endpoint, cells are either fixed for immunocytochemistry/immunofluorescence or analyzed using TFM. Scale bars are 75 µm. Please click here to view a larger version of this figure.
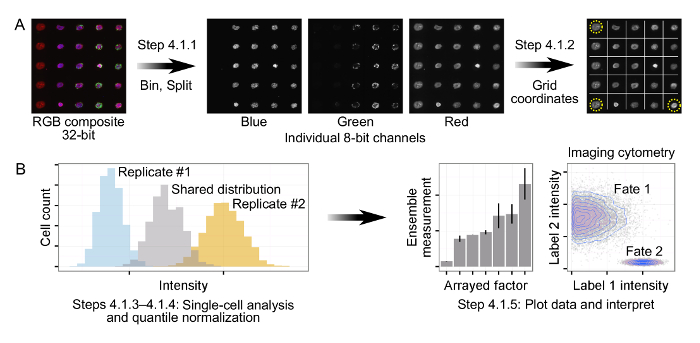
Figure 2: Processing and Analysis of Immunofluorescence Data from Arrays. (A) Tiled, composite 32-bit RGB images are first binned and then split into individual 8-bit channels. Using a combination of arrayed fluorescent markers and cell islands, three corners of the array are identified to allow for automated orientation and gridding of the arrays. (B) Single-cell data is generated for each channel of the input arrays. In order to account for experimental drift, quantile normalization is applied by biological replicate, producing a single shared distribution across all replicates. Quantile normalized data is subsequently plotted and interpreted via calculation of ensemble measurements (e.g., cells/island, mean intensity, percentage cells positive for a label) or direct analysis of single-cell distributions. Please click here to view a larger version of this figure.
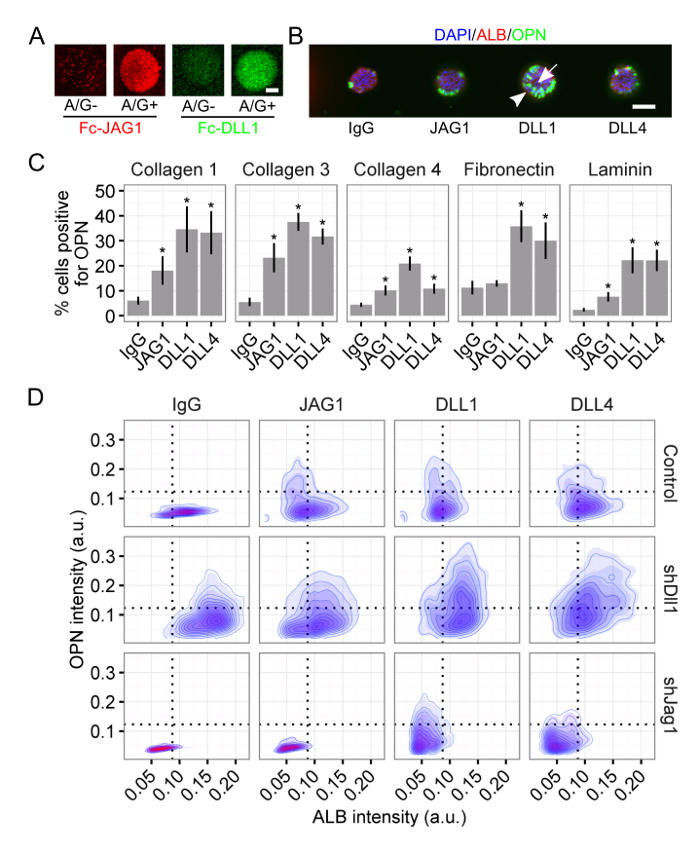
Figure 3: Notch Ligand Presentation Mediates Liver Progenitor Differentiation. (A) Fc-recombinant Notch ligands Jagged-1 (JAG1) and Delta-like 1 (DLL1) exhibited improved retention and clustering when arrayed with Protein A/G. Scale bar is 50 µm. (B) Liver progenitors differentiated into bile duct cells upon presentation with Notch ligand. 4',6-Diamidino-2-phenylindole (DAPI) is a nuclear label, albumin (ALB) is a hepatic cell marker, and osteopontin (OPN) is a bile duct cell marker. Scale bar is 150 µm. (C) Quantification of percentage of cells positive for OPN for the Notch ligands JAG1, DLL1, and Delta-like 4 (DLL4) on the ECM proteins collagen I, collagen III, collagen IV, fibronectin, and laminin. Student's t-tests were performed against control IgG for each arrayed Notch ligand within each ECM protein with P-values indicated for P<0.05 (*). (D) Imaging cytometry of ALB and OPN for cells on collagen III presented with the Notch ligands JAG1, DLL1, and DLL4. Liver progenitors without the Notch ligands Dll1 and Jag1 (i.e., shDll1 and shJag1) were generated using shRNA knockdown. Data in (C) presented as mean ± s.e.m. This figure has been modified from Kaylan et al.34. Please click here to view a larger version of this figure.
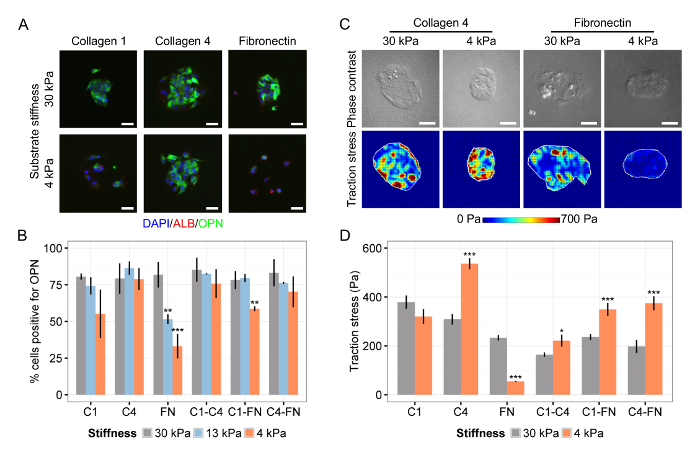
Figure 4: Matrix Composition and Substrate Stiffness Coordinate Liver Progenitor Differentiation. (A) Liver progenitor differentiation to bile duct cells is dependent on both ECM composition and substrate stiffness. DAPI is a nuclear label, ALB is a hepatic cell marker, and OPN is a bile duct cell marker. (B) Quantification of percentage of cells positive for OPN on substrates of Young's modulus 30 kPa, 13 kPa, and 4 kPa for collagen I (C1), collagen IV (C4), fibronectin (FN), and all two-way combinations of those ECM proteins. (C) Cell traction stress is dependent on both substrate stiffness and ECM composition. (D) Quantification of root-mean-square values of traction stress on substrates of Young's modulus 30 kPa and 4 kPa for collagen I (C1), collagen IV (C4), fibronectin (FN), and all two-way combinations of those ECM proteins. In (B) and (D), data were presented as mean ± s.e.m and Student's t-tests were performed against 30 kPa for each ECM combination with P-values indicated for P< 0.05 (*), P< 0.01 (**), and P< 0.001 (***). Scale bars are 50 µm. This figure has been modified from Kourouklis et al.35. Please click here to view a larger version of this figure.
| Section | Problem | Potential Causes | Solution |
| 1. Fabrication of Polyacrylamide Substrate. | Coverglass cannot be removed from hydrogel. | Overpolymerization. | Reduce polymerization time to <10 minutes (4 W/m2). Check that UV crosslinker output is within expected range. |
| Poor polyacrylamide hydrogel polymerization. | Underpolymerization. | Increase polymerization time to >10 minutes (4 W/m2). Check that UV crosslinker output is within expected range. | |
| Polyacrylamide hydrogels are damaged after removal of coverglass. | Soft polyacrylamide hydrogels are easy to damage. | We observe decreasing hydrogel fabrication yield (~50%) for the softest (i.e., 4 kPa) hydrogels in particular. Handle hydrogels gently and increase starting numbers to attain desired yield. | |
| 2. Fabrication of Arrays. | Poor or inconsistent spot morphology. | Inconsistent humidifier function. | Check that humidifier and rheometer a functional throughout each print run and maintain 65% RH. |
| Pins stuck in printhead or clogged. | Clean the printhead to allow for free pin movement. Clean pins thoroughly before or after each print run to remove aggregates from pin channels. | ||
| 3. Cell Culture and Assay Execution. | Cell detachment or death on arrays after initial attachment. | Overseeding and excessive proliferation. | Reduce initial seeding density and time. Use "maintenance" or "differentiation" media during array culture to reduce cell proliferation. |
| Release of toxic acrylamide monomer from hydrogel. | Soak hydrogels in dH2O for at least 3 d to allow for diffusion/release of acrylamide monomer and reduce cell toxicity. | ||
| Cells don't attach to arrays. | Underseeding. | Increase initial seeding density and time. Use a more strongly adherent cell type. | |
| Poor deposition of matrix or biomolecule condition. | Clean pins of particles and aggregates, confirm printing parameters, and evaluate spotting of fluorescent markers, e.g., rhodamine-conjugated dextran. | ||
| Specificity of cell–matrix interactions. | Different cell types adhere specifically to some but not other ECM proteins. Test multiple different ECM proteins with your cells. | ||
| Suboptimal array storage after fabrication. | We recommend storing fabricated arrays overnight at 65% RH and room temperature, in part to avoid phase changes during freezing. Cell adhesion is sensitive to both humidity, temperature, and storage time; make sure these parameters are consistent/optimized for your experiments. | ||
| Detachment of hydrogel from glass substrate during cell culture. | Poor slide cleaning and silanization. | Replace working solutions for slide cleaning and silanization. | |
| Overdehydrated hydrogel. | Don't leave hydrogels dehydrating on a hot plate for longer than 15–30 min. | ||
| 4. Analysis of Data. | High variability between replicate spots and slides. | Variability in array fabrication. | Check that pins and printhead are clean. Confirm humidifier function. Visualize and quantify spot and array quality using fluorescent markers. Store arrays as recommended above. |
Table 1: Troubleshooting.
