In this section, we present representative results that were obtained following the observation, at the transmission EM level, of immunostained primate brain tissue chemically fixed with a mixture of 3% acrolein and 4% PFA. We achieved good preservation of the ultrastructure, as indicated by the relatively intact myelin sheath and the neat visualization of double membranes (Figure 2A). Synaptic contacts, along with neuronal elements from the microenvironment, can easily be identified (Figure 2B). Neuronal elements labeled with diaminobenzidine (DAB) immunoprecipitate are recognized at the EM level by their filled cytoplasm or axoplasm. The plasma membrane and the outer surface of organelles are also typically lined with the electron-dense precipitate (Figure 3).
In this particular experiment, we used antibodies against the serotonin transporter (SERT), choline acetyltransferase (ChAT), or Tyrosine Hydroxylase (TH) to visualize immunolabeled neuronal elements in the external (GPe) or internal (GPi) segment of the squirrel monkey globus pallidus (Figure 3). In order to do so, we used a combination of fixative chemicals that preserve antigenicity as well as ultrastructure, allowing a detailed morphological investigation. Although many antibodies can be used with the transcardiac perfusion protocol described above, we recommend that users perform optimization concentration tests beforehand, since some primary antibodies are known to not provide optimal immunolabeling with acrolein fixation. Alternatively, when antibodies do not provide optimal immunolabeling with acrolein fixation, a dilution of 0.1 – 2% glutaraldehyde in 4% PFA can be used for transcardiac perfusion. It provides tissue quality relatively equivalent to acrolein-fixed brain tissue with preserved antigenicity for many antibodies (Figure 4).
Finally, we provide typical examples of EM photomicrographs obtained following inappropriate manipulations. A poor fixation results in altered myelin sheaths (Figure 5A, B) and difficulties in visualization of the double membranes of neurites (Figure 5C, D), preventing a reliable identification and analysis of labeled and unlabeled neuronal elements. An excessive incubation time in the DAB solution creates excessive background and non-specific staining that can potentially generate false-positive results. Background or non-specific staining sometimes appears as incomplete staining of neuronal elements (Figure 6A, B), but more often as numerous and closely located stained neuronal elements (Figure 6C, D). The use of detergent in the blocking solution significantly alters the quality of the tissue. It may lead to missing organelles in labeled elements (Figure 7A) or degradation of myelin sheaths (Figure 7B) and cellular membranes (Figure 7C), rendering any acute interpretation of the microenvironment difficult. Finally, a misstep in the osmification process, such as using rinsing solution containing sodium chloride, produces unreliable results where the cellular structures are difficult to distinguish (Figure 7D).
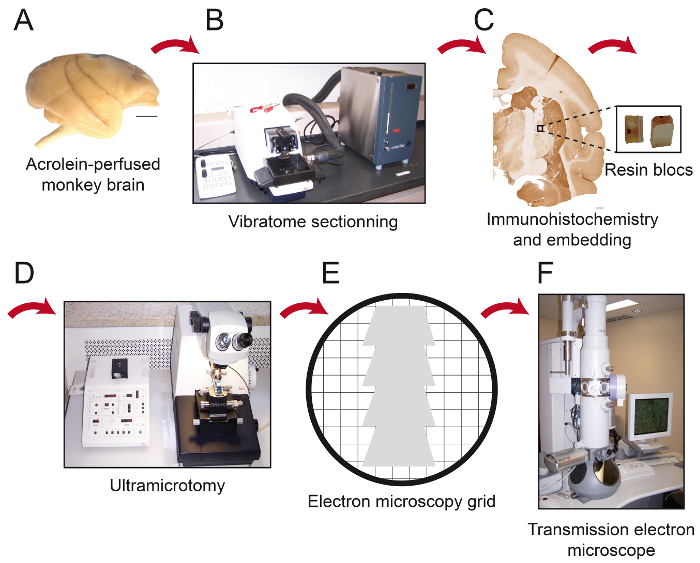
Figure 1: Schematics of Essential Steps of the Protocol. The monkey brain (A) is cut into serial sections with a cooling vibratome (B). It is then processed for pre-embedding immunohistochemistry and electron microscopy, after which the region of interest is placed on the tip of a resin block (C) and cut into 80 nm thick sections with an ultramicrotome (D). Ultrathin sections are then collected on bare 150 mesh copper grids or formvar-coated nickel grids (E), stained with lead citrate and ready to be observed under transmission electron microscope (F). Scale bars: 1 mm. Please click here to view a larger version of this figure.
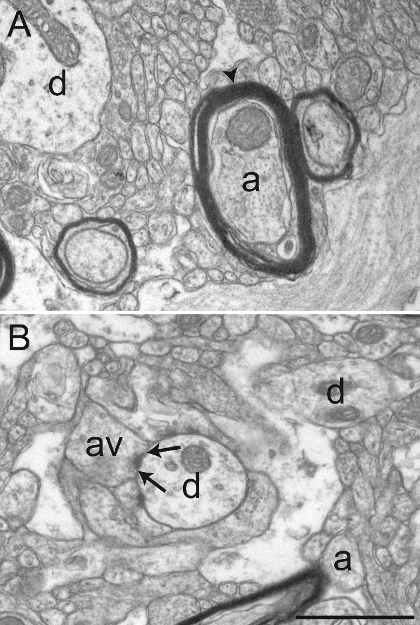
Figure 2: Well-preserved Primate Brain Tissue after Acrolein-PFA Transcardiac Perfusion. Electron micrographs of squirrel monkey (Saimiri sciureus) brain tissue of the GPi (A) and GPe (B) showing representative well-preserved material after performing the acrolein-PFA transcardiac perfusion and the immunoperoxidase-diaminobenzidine technique. The myelin sheath of axons (a) is relatively intact (see arrowhead in A), and the general ultrastructure is well preserved in A and B. Dendritic profiles (d), small unmyelinated axons (a), and axon varicosities (av) can easily be identified. An example of an axon varicosity establishing a symmetrical synaptic contact (between arrows) with a dendritic profile (d) is shown in B. Scale bar: 1 µm. Please click here to view a larger version of this figure.
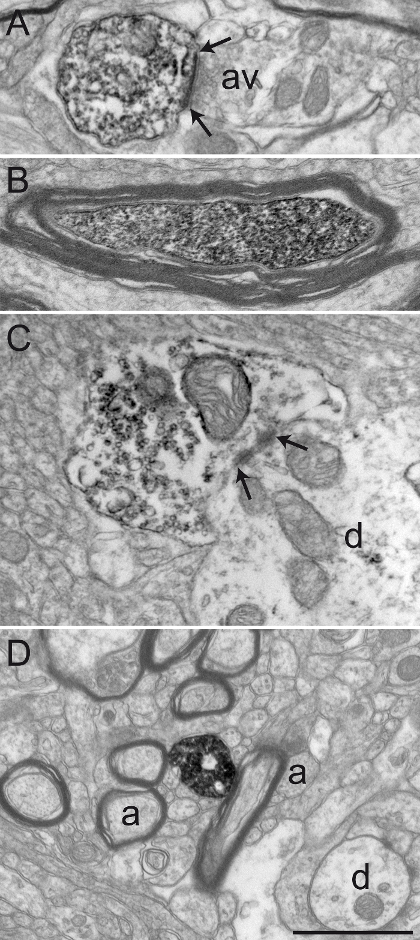
Figure 3: Sections from Squirrel Monkey GPe and GPi Immunolabeled for Choline Acetyltransferase (ChAT), Serotonin Transporter (SERT), and Tyrosine Hydroxylase (TH) by using the Immunoperoxidase-diaminobenzidine Technique. Immunolabeled elements can easily be identified by their cytoplasm or axoplasm filled with electron-dense DAB precipitate. Labeled dendritic profiles are recognized by filled microfilaments, as seen in A, where a ChAT-immunostained dendrite in the GPi receives a synaptic contact (between arrows) from an unlabeled axon varicosity (av). The electron micrograph in B shows a myelinated axon in the GPe with a relatively intact myelin sheath whose axoplasm is immunolabeled for TH. The example in C shows an axon varicosity in the GPi immunolabeled for SERT and is seen to establish a symmetrical synaptic contact (between arrows) with a dendrite (d). In this example, the DAB precipitate lines the plasma membrane and the outer surface of organelles (mitochondrion and synaptic vesicles). The axon varicosity shown in D was observed in the GPe and immunolabeled for TH and represents an example of DAB precipitate entirely filling the axoplasm, with synaptic vesicles being visible but more difficult to delineate. Elements of the microenvironment can easily be identified, as exemplified by myelinated and unmyelinated axons (a) and occasional dendrites (d) surrounding the labeled axon varicosity. Electron micrographs are modified from 25,26,27. Scale bar: 1 µm. Please click here to view a larger version of this figure.
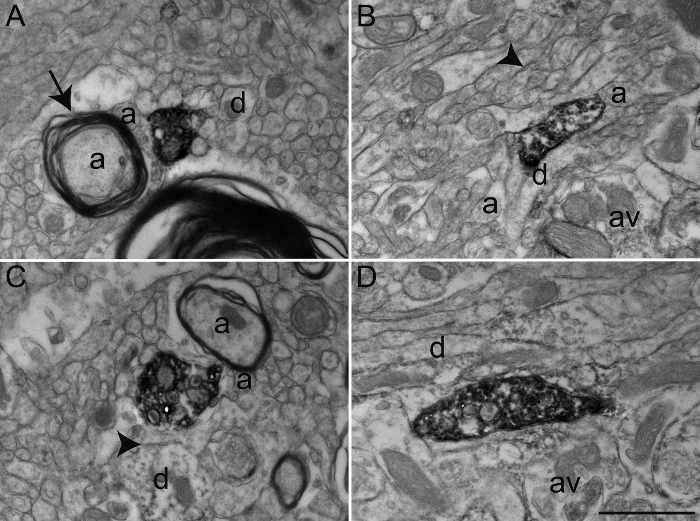
Figure 4: Examples of Primate Brain Tissue after Glutaraldehyde-PFA Transcardiac Perfusion. Representative electron micrographs of macaque monkey (Macaca fascicularis) brain tissue of the GPe (A) and GPi (B-D) after performing transcardiac perfusion with 0.2% glutaraldehyde mixed with 4% PFA and immunoperoxidase-diaminobenzidine technique with an antibody against the serotonin transporter (SERT). As in Figure 2, immunolabeled elements can be identified by their cytoplasm and axoplasm filled with electron-dense DAB precipitate. General ultrastructure is relatively intact and elements of the microenvironment can easily be identified, as shown by myelinated and unmyelinated axons (a) and occasional dendrites (d) and axon varicosities (av) surrounding the labeled axon varicosities, as described in Figure 2. However, note the inconsistency in the quality of ultrastructure, denoted by well-defined plasma membranes (arrowheads), but relatively damaged myelin sheath (arrow). Scale bar: 1 µm. Please click here to view a larger version of this figure.
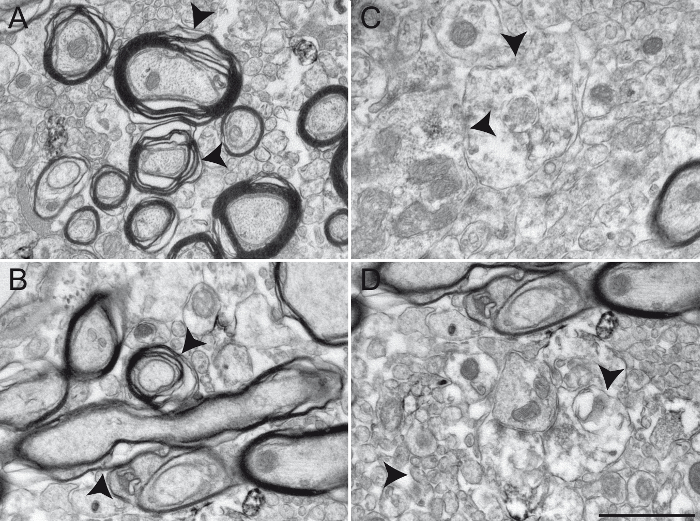
Figure 5: Examples of Primate Brain Tissue Obtained following an Unsuccessful Transcardiac Perfusion. Results from an unsuccessful chemical fixation are shown here in the squirrel monkey GPi (A - B) and GPe (C - D) transcardially perfused with 0.9% NaCl rinsing solution and a mixture of ice-cold 4% PFA and 15% picric acid diluted in 0.1 M PB (pH 7.4). Brains were post-fixed 1 h at 4 °C in 4% PFA and 30% sucrose and cut into 60 µm thick sagittal sections with a cooling vibratome. Inappropriately fixed brain tissue can be recognized by a damaged myelin sheath (A and B, arrowheads), as well as by blurry or undefined plasma membranes (C and D, see arrowheads for examples). Different neuronal elements are difficult to identify, rendering any interpretation of the ultrastructure unreliable. Scale bar: 1 µm. Please click here to view a larger version of this figure.
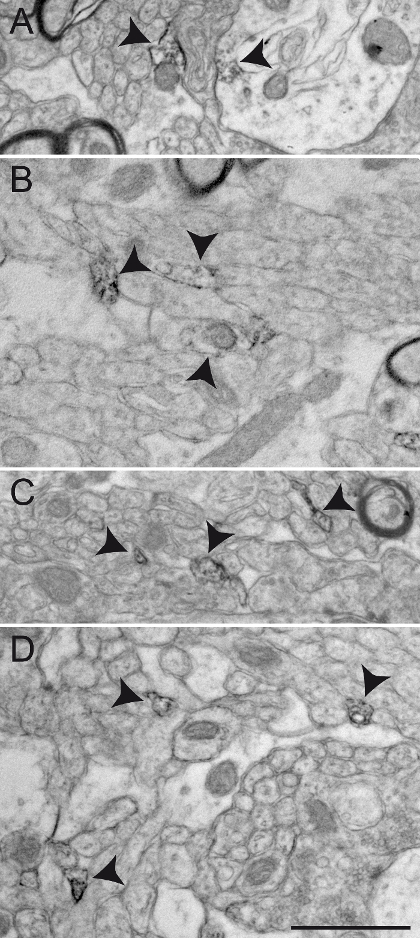
Figure 6: Examples of Non-specific ChAT Immunolabeling in the Squirrel Monkey GPe. Background staining or non-specific immunolabeling sometimes appears under the EM as partial staining of large cellular elements, as illustrated in A - B (arrowheads). Such unspecific staining appears more often at the surface of immunostained sections. Other examples of non-specific immunolabeling include the frequent observation of very small elements evoking small, unmyelinated axons partially or completely filled with DAB and located very close to one another, as demonstrated by arrowheads in C and D. Scale bar: 1 µm. Please click here to view a larger version of this figure.
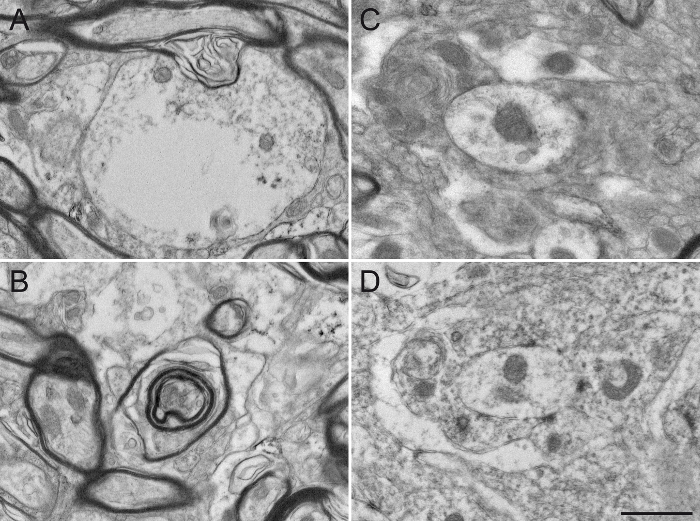
Figure 7: Damaged Squirrel Monkey GPe Tissue after Missteps in Sample Preparation for Electron Microscopy. The use of detergent, such as Triton X-100, in the blocking solution, even at a low concentration of 0.02%, substantially alters the integrity of the ultrastructure by damaging the cytoplasm of dendrites (A) or the myelin sheath of axons (B). The different neuronal elements are also difficult to distinguish from one another (C), since the plasma membranes are damaged and difficult to delineate. The osmification process is also an important step in sample preparation. The use of sodium chloride in rinsing solutions (D) alters the fixation of the tissue, rendering subsequent analysis difficult. Scale bar: 800 nm. Please click here to view a larger version of this figure.
