1. Functionalization of CNTs and GNPs by Ultrasonic Ozonolysis
- Weigh the nanomaterials in a glove box inside a HEPA filter-equipped fume cupboard. Weigh the desired quantity of nanomaterials into a beaker. Transfer to a bottle and add ultrapure water to make a concentration of 1 g/L.
- Seal the bottle with a lid. Ultrasonicate in a standard ultrasonic bath (see Materials List; frequency: ~43 ±2 kHz; power: 60 W) to disperse the CNTs or GNPs.
NOTE: CAUTION. See the Work Health and Safety Notice above. - Carefully pour the nanomaterial suspension into a reactor flask containing a magnetic stirrer bar. Attach a reactor flask lid with sufficient ports for the ozone and ultrasonic horn inlets and outlets. Place this on a stirring plate and turn on the magnetic stirrer by adjusting the dial to prevent the CNTs from settling out of suspension.
- Assemble the ultrasonic horn into the ultrasonic cell and check that the horn tip is in good condition. See Figure 1.
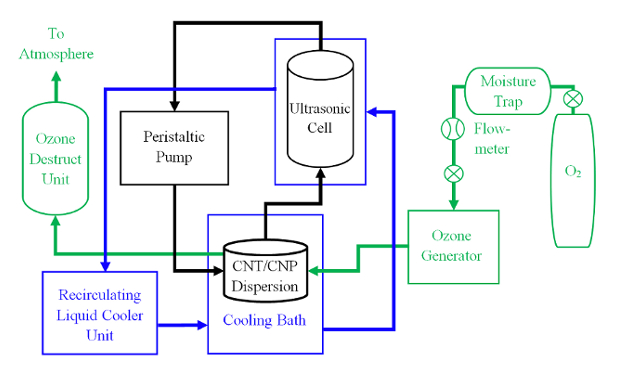
Figure 1: Ultrasonic Ozonolysis System. This schematic diagram illustrates how to connect the various elements of the ultrasonic ozonolysis system. Please click here to view a larger version of this figure.
- Use silicone tubing to connect the reactor, the ultrasonic cell, and the peristaltic pump. Refer to Figure 1.
- Insert the ozone bubbler into the reactor and connect it to the ozone generator.
NOTE: CAUTION. See the Work Health and Safety Notice above. - Connect the recirculating liquid cooler unit to a cooling coil (to cool the reactor) and to the ultrasonic cell-cooling jacket. Flip the switch to turn on the recirculating liquid cooling unit to cool the ozone reactor and ultrasonic cell. Ensure that the cooling liquid is flowing through both the ultrasonic cell and the reactor cooling bath.
- Use a thermocouple connected to the ultrasonic horn control unit to measure the reactor temperature. Wait until the system has cooled to 5 °C.
- Place the reactor flask with the CNTs in position. Insert the ozone diffuser into the bottom of the reactor flask so that it is fully submerged. Ensure that the outlet tube to the ultrasonic cell is submerged at least 25 mm below the surface of the dispersion.
- Connect the outlet tube from the reactor to the ozone destruct unit using the same silicone tubing as in the peristaltic pumping operation. Use of a water bubbler on the outlet side of the ozone destruct unit is optional, but it helps to capture any residual ozone and provides an indication that the ozone generator is connected and working correctly.
- Turn on the oxygen supply to the ozone generator and adjust the valve to achieve a 0.5-L/min flow rate through the ozone generator.
- Turn on the ozone generator by flipping the switch. Allow it to run for 30-60 min prior to starting the pumping or the ultrasonication process.
- Turn on the peristaltic pump, adjust it to 0.67 Hz by turning the pump dial to a setting of approximately 5 or 6, and ensure that the CNT dispersion flows evenly through the cell.
- Turn on the ultrasonic horn and adjust the power to 60 W by entering this value into the control module under the manual operation menu.
- At initial startup, ramp up the amplitude setting slowly to achieve the desired power output of 60 W.
NOTE: The ultrasonic horn operates at 20 kHz. The amplitude setting and power setting are usually of similar values, but upon initial startup, the amplitude setting may need to be slowly increased to achieve the desired power output of 60 W.
- Observe the sonication process for at least 30 min to ensure stable pumping and ultrasonic horn operation.
- Monitor the flexible tubing in direct contact with the peristaltic pump mechanism for wear. Adjust the tube position at least every 2 h to ensure that the tubing maintains integrity. Replace the tubing for each separate carbon nanomaterial processing run.
- When the desired USO processing time has elapsed, press the switches to turn off the ozone generator, sonicator, and pump. Maintain stirring until the carbon nanomaterial dispersion is ready to be transferred to a sealed container for later use in electrophoretic deposition.
- Wait around 1 h before transferring the dispersion to allow the ozone in solution to decompose.
- If electrophoresis is not performed within 24 h of USO processing, sonicate the nanomaterial dispersion again to minimize the number of particles settling out of dispersion. Insert the sonic horn into the bottle of the carbon nanomaterial dispersion and sonicate (as above) for 30 min prior to use.
2. Electrophoresis
- Prepare three stainless-steel electrodes, 60 mm (L) x 25 mm (W), from 0.2-mm sheet material.
- Abrade the electrodes using P1000 aluminum oxide sandpaper with ultrapure water as a lubricant. Following the abrasion, place the electrodes in the ultrasonic cleaning bath and clean them for 10 min in ultrapure water.
- Place the electrode to be used for anodic deposition in a clean oven at 100 °C and dry it for 10 min. Move the electrode to a desiccator and allow it to cool. Weigh the electrode on a four-figure analytical balance.
- Record the weight of the electrode to four decimal places as the uncoated weight in grams. Dry the remaining two electrodes and store them in the desiccator for use as cathodes for deposition.
- Using a suitable clamping arrangement (see Figure 2), such as a small vice and 10-mm non-conductive plastic spacers, assemble the electrodes, with the outer electrodes being used as the cathodes and the inner electrode as the anode.
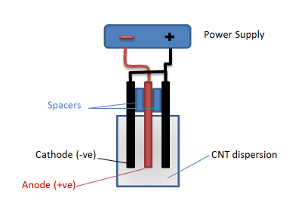
Figure 2: Electrophoretic Deposition Cell. This schematic diagram illustrates the configuration of the electrophoretic deposition cell. Please click here to view a larger version of this figure.
- Transfer 35 mL of carbon nanomaterial dispersion to a 50-mL beaker.
- Place the stainless-steel electrode fixture onto a retort stand and slowly lower the electrodes to the full depth of beaker. Attach the positive terminal of the DC power supply to the central stainless-steel anode. Attach the negative terminal of the DC power supply to the outer cathode. Connect the two outer cathodes using a lead with alligator clips.
- If accurate current measurements are required, connect a multimeter in series with the power supply unit and electrodes to enable monitoring during deposition.
- Manually adjust the current setting on the DC power supply to the maximum current (nominally, 2 A) and then the voltage to the required value for the studies, ensuring that the power supply output is turned off. Prepare a stopwatch and switch on the power supply for the required coating duration.
- For a typical coating experiment, adjust the voltage to 10 V, with the coating times ranging from 1 to 15 min. Switch off the DC power supply and then slowly raise the electrodes out of the dispersion, ensuring that the film is not disturbed.
- Disconnect the terminals from the electrodes and slowly invert the electrodes to a horizontal orientation to allow the film on the anode to dry evenly. After the moisture has evaporated from the film at room temperature, disassemble the electrode fixture and place the anode in the oven at 100 °C to dry for 1 h.
- Clamp and hang the anode in the oven to ensure that the film does not come into direct contact with any surfaces.
- Place the film-coated anode in the desiccator and allow it to cool, then weigh it on a four-figure analytical balance. Record the weight to four decimal places. Subtract the weight of the uncoated anode to determine the mass of the deposited film.
- Photograph the coated anode. Use a suitable image processing package to accurately measure the area of the deposited film. Use the mass and area to record the areal density of the film in mg/cm2.
- Repeat the coating and weighing steps for each deposition time required to determine the rate of deposition of the carbon nanomaterial onto the stainless steel for the specific field strengths being used.
3. Chemical Characterization – X-ray Photoelectron Spectroscopy (XPS)15
- To prepare the samples for XPS analysis, drop-cast the aqueous CNT dispersions from the original 1 g/L dispersion.
- Using a pipette, deposit a droplet of the dispersion on a stainless-steel disk and it allow to dry at room temperature. Repeat the process until a uniformly thick CNT coating of approximately 2 µm in thickness is observed on the disk. Mount the stainless-steel disk onto sample stubs using double-sided conductive tape.
- Insert the samples into the sample entry chamber and pump down to a vacuum of 5 x 10-7 Torr prior to transferring it to the main analysis chamber. Wait for the main chamber pressure to reach 5 x 10-9 Torr before positioning the sample and undertaking the analysis.
- To follow the typical operating conditions for the XPS measurements, use a monochromatic Al Kα,1 X-ray source operating at 150 W to illuminate an area of 700 µm x 300 µm. Detect photoelectrons at a 90° take-off angle with respect to the sample surface.
- Calibrate the spectrometer energy scale using the Au 4f7/2 photoelectron peak at a binding energy of 83.98 eV.
NOTE: The energy scale is adjusted in the instrument control software during the initial commissioning of the instrument. It is regularly checked following any maintenance operations by the spectroscopist in charge of the equipment and should not be altered by users.
- Calibrate the spectrometer energy scale using the Au 4f7/2 photoelectron peak at a binding energy of 83.98 eV.
- Monitor the initial sample charge by positioning a 30-eV window over the C 1s region. Perform charge compensation by adjusting the electron flood gun parameters systematically to position the C 1s peak at 284.6 eV and to minimize the peak width at half the maximum peak intensity (FWHM).
- Conduct survey scans at 160 eV pass energy and 0.5 eV steps, with a dwell time of 0.1 s, in fixed analyzer transmission mode to identify the surface elemental species. Acquire region spectra at a 20 eV pass energy using 0.05-eV steps and a 0.2 s dwell. Use between 4 and 8 sweeps to improve the signal-to-noise ratio.
- To quantify the spectra, use a Shirley background fit and sensitivity factors from a suitable elemental library16.
- Deconvolve C 1s high-resolution X-ray photoelectron spectra to determine the different carbon-oxygen species present with USO treatment time15.
- To accurately deconvolute the C 1s peak, perform an initial peak fit to the shape of graphitic carbon to ensure that the peak shape due to the sp2 bonding and the contributions from the low-energy π-type shake-up satellites are maintained17. For all peak fitting, constrain the component binding energies and FWHM by ±0.1 eV and ±0.2 eV, respectively, and use a Gaussian/Lorentzian ratio of 30.
- Fit the graphite peak shape using six separate peaks. Maintain at fixed ratios the areas of each peak relative to the main graphite peak at 284.6 eV to maintain a consistent shape due to the graphitic component in the processed nanomaterial15,9,14.
4. Structural Characterization – Raman Spectroscopy18
- Prepare the nanomaterials for Raman spectroscopic characterization by diluting the original dispersion with deionized water to achieve a 0.1 g/L concentration. Pipette 0.05 mL of dispersion onto a suitable polished-gold substrate. Allow the droplet to evaporate at room temperature to produce a thin carbon film on which to focus the Raman laser spot.
NOTE: The size of the gold substrate is incidental to the result when a large substrate is used, multiple specimens can be deposited on a single slide. - Undertake Raman measurements with a 50X magnification objective lens using a dispersive confocal microscope and a 532 nm laser. Acquire the spectra between 3,500 and 50 cm-1 at a resolution of 2 cm-1. Use a 50 µm pinhole aperture with a laser energy between 1 and 10 mW, using the maximum energy possible such that no sample degradation occurs during collection.
- Collect the spectra by accumulating between 10 and 50 scans and using an exposure time between 2 and 5 s for each individual scan to achieve a reasonable signal-to-noise ratio.
5. Film Morphology – Scanning Electron Microscopy (SEM)
- For the SEM analysis of functionalized versus unfunctionalized CNT/GNPs, dilute a droplet of each dispersion to 0.1 g/L using deionized water. Deposit it onto an aluminum SEM stub and allow to dry in air overnight.
- For EPD films deposited onto the stainless-steel electrodes, cut the electrodes into 1 cm x 1 cm squares and mount them onto sample stubs using double-sided conductive tape. Paint a small line of silver dag (i.e., conductive adhesive/paint) from the sample stub onto the top surface of the specimen to improve the conductive path. Dry the sample under a heater lamp for at least 15 min.
- Place the mounted specimen in the sputter coater and give it a conductive iridium coating approximately 1.3-1.5 nm thick19.
- On the thickness measurement unit, set the material density to the appropriate level, which is 22.56 g/cm3 for iridium. Adjust the target thickness setting to 1.0 nm to give a thickness of approximately 1.3-1.5 nm when accounting for thermal drift during the coating process.
- Set the coater unit to allow the sputter coater operation to be controlled by the thickness measurement unit. Select the desired power output, which should be 80 mA for a high-resolution coating19.
- Zero the coating thickness gauge on the thickness control unit and press the "Cycle" button to commence the automated chamber evacuation, argon bleed/flush and coating sequence. When the power has reached the desired level, move aside the target shield to allow the coating to commence. Rotate and tilt the stage during the coating to facilitate an even coating on all sides.
- Examine the nanomaterials using a field-emission SEM with an in-lens detector. Use a working distance of approximately 3 mm and an accelerating voltage of 3.0 kV.
Figure 3 shows the XPS wide-scan characterization of CNTs that had undergone USO treatment. CNTs that had not undergone USO show almost no oxygen content. As the USO time increases, the surface oxygen level increases. Figure 4 charts the oxygen-to-carbon ratio increases as a function of USO time. Table 1 shows the deconvoluted carbon species atomic concentrations of GNP treated with USO. The peak fitting used a combination of constrained peaks, represented by Gr1 to Gr6, to represent the inherent peak shape due to graphite and the associated energy loss features17. The oxygen-containing species were then added, and a cross-correlation with the C 1s peak fitting data and the elemental C and O percentages was made to ensure that sensible peak fitting results were achieved. The C-O, C=O, and COO species are at insignificant levels without USO treatment but increased markedly with 12 h of USO.
Raman spectra of CNTs treated by USO are shown in Figure 5. The intensity of the D band at 1,346 cm-1 denotes the presence of defects in the CNTs20 and indicates that defects already existed in the CNTs prior to the USO treatment. With increasing USO treatment time, there is a shift in the G band, from 1,576 cm-1 to 1,582 cm-1, with a second component becoming more pronounced at 1,618 cm-1. This corresponds to an increase in the oxidation level of the CNTs. There is also a noticeable decrease in intensity of the 2D band at 2,698 cm-1 and a small increase in the D+G band at 2,941 cm-1 with USO treatment time, indicating an increased level of oxidation in the CNTs20.
The intensity ratio of the D to G bands, ID/IG, was measured at 1.18 with no USO, increased to 1.37 after 155 min of USO treatment, and continued to increase at a much slower rate with greater USO treatment times. This may indicate that oxidation is occurring primarily at existing defects, and once the existing defect sites are saturated, the oxidizing effect of further USO treatment time occurs at a slower rate. This suggests that there is minimal damage to the as-received CNT structure from USO.
The SEM images in Figure 6 show USO-treated CNTs compared with untreated CNTs. The untreated CNTs are highly clumped, while the USO-treated CNTs are much more evenly dispersed across the surface, showing that the USO has helped to reduce agglomerates present in the as-received material.
The USO-treated CNTs were then electrophoretically deposited using a variety of conditions. Thicker films were produced as the electrophoresis time increased; thicker films were also produced by using higher CNT concentrations, as shown in Figure 7. Similar results are achieved when GNPs are used. Figure 8 shows the rate of electrophoretic deposition at various CNT concentrations, using deposition rates calculated from curve-fitting the data in Figure 7. It appears that the rate of EPD of USO-treated CNTs can be predicted reasonably well if the initial CNT concentration is known. The linear relationship in deposition rate and dispersion concentration is predicted from the Hamaker Law21, but the logarithmic rate in deposition shows that changes in the deposition parameters are occurring with time. Our previous work, in which CNTs were deposited onto carbon12 and glass13 fabrics, also clearly showed that the deposition rate with time is non-linear. Under constant voltage deposition, the resistance of the CNT film increases, leading to a reduction in the deposition rate.
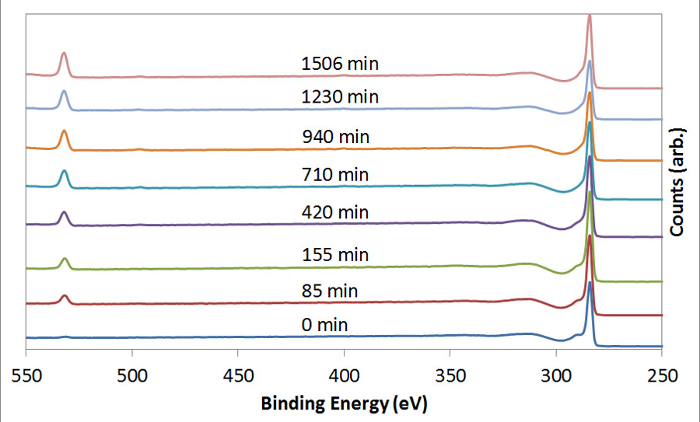
Figure 3: Chemical Characterization – XPS. CNTs that had been subjected to USO for varying times were analyzed using XPS. This image shows the wide scan changes in oxygen (532 eV) and carbon (284.6 eV) intensity, with the oxygen intensity increasing at higher USO times. Please click here to view a larger version of this figure.
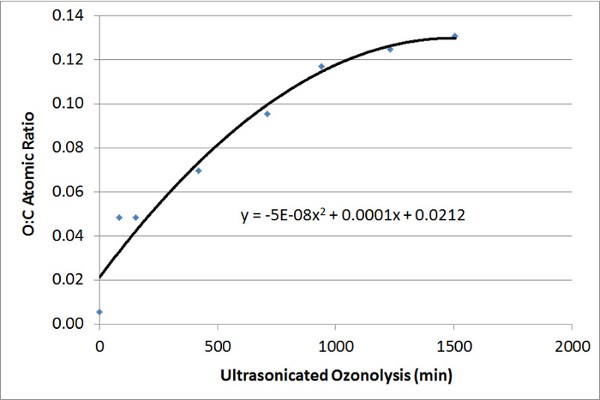
Figure 4: Atomic Ratio – XPS. This graph shows the change in the O:C atomic ratio of CNTs with increasing USO times, calculated from the scans shown in Figure 3. The ozonolysis mechanism is very complex, and the polynomial fit is provided, as a guide to the reader, to show the trend in oxidation rate, but not to explain anything about the ozonolysis mechanism. Please click here to view a larger version of this figure.
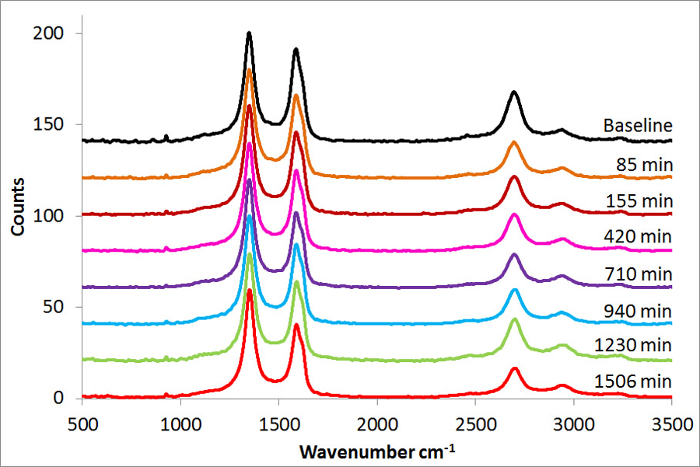
Figure 5: Structural Characterization – Raman. All spectra showed strong D bands (~1,346 cm-1), G bands (~1,576 cm-1), 2D bands (~2,698 cm-1), and D+G bands (~2,941 cm-1). The D-band increase indicates some increase in the graphitic defects. Please click here to view a larger version of this figure.
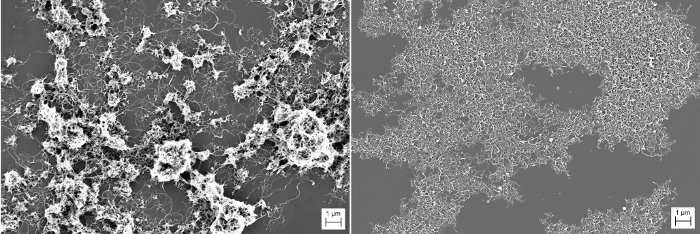
Figure 6: Film Morphology – SEM. These images show CNTs that were (a) not chemically treated and (b) USO-treated for 16 h. Please click here to view a larger version of this figure.
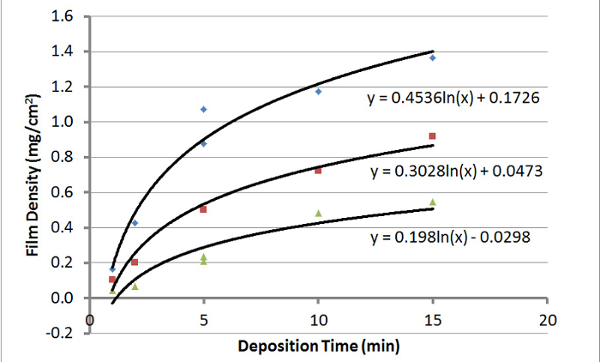
Figure 7: Electrophoresis Measurements. This graph shows the density of the EPD films versus the deposition time using three different concentrations of CNTs dispersed in water. Blue diamonds = 2 g/L, red squares = 1.5 g/L, and green triangles = 1 g/L. A 14 V/cm DC electric field was used for these samples. Please click here to view a larger version of this figure.
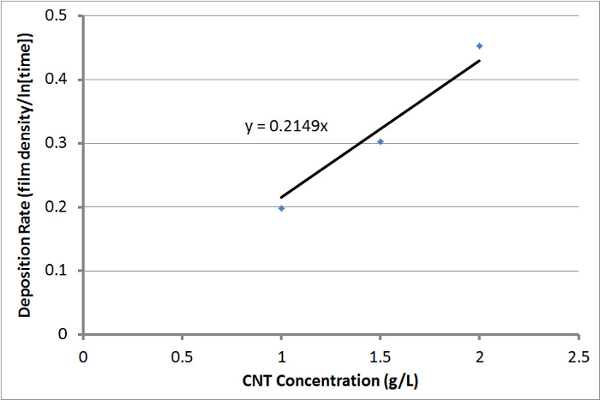
Figure 8: Electrophoretic Deposition Rate. Plot of the EPD rate versus the CNT concentrations, using deposition rates calculated from the curve fit of the Figure 7 plot of film density versus deposition time. Please click here to view a larger version of this figure.
| Name | Posição | Constraint | FWHM | Constraint | Area Constraint | % Atomic Concentration | |
| 0 h | 12 h | ||||||
| Gr 1 | 284.6 | ±0.1 | 0.7 | ±0.2 | Best fit | 29.4 | 17.6 |
| Gr 2 | 284.8 | 1.2 | Gr1*1.3 | 36.9 | 22.2 | ||
| Gr 3 | 286.1 | 1.3 | Gr1*0.4 | 12.5 | 7.5 | ||
| Gr 4 | 287.9 | 1.3 | Gr1*0.2 | 5.9 | 3.5 | ||
| Gr 5 | 289.5 | 1.6 | Gr1*0.1 | 3.3 | 1.8 | ||
| Gr 6 | 291.3 | 2.4 | Gr1*0.2 | 6.6 | 1.7 | ||
| C-C | 285 | 1.2 | Best fit | 2.1 | 13.7 | ||
| C-O | 287 | 1.2 | Best fit | 1.5 | 14.3 | ||
| C=O | 288.6 | 1.2 | Best fit | 0.9 | 12.4 | ||
| COO | 289.4 | 1.2 | Best fit | 0.9 | 5.3 | ||
Table 1: Chemical Characterization – XPS. This table shows the peak fitting parameters used to deconvolute the C 1s spectrum for the GNPs processed using the USO method for 0 h and 12 h treatment times. It also shows the resulting atomic concentrations of each species14. Gr 1, Gr 2, etc. represent the various graphite peaks used in the fit.
