Dissection and Culture of Mouse Embryonic Kidney
Summary
This protocol describes a method for isolating and culturing metanephric rudiments from mouse embryos.
Abstract
The goal of this protocol is to describe a method for the dissection, isolation, and culture of mouse metanephric rudiments.
During mammalian kidney development, the two progenitor tissues, the ureteric bud and the metanephric mesenchyme, communicate and reciprocally induce cellular mechanisms to eventually form the collecting system and the nephrons of the kidney. As mammalian embryos grow intrauterine and therefore are inaccessible to the observer, an organ culture has been developed. With this method, it is possible to study epithelial-mesenchymal interactions and cellular behavior during kidney organogenesis. Furthermore, the origin of congenital kidney and urogenital tract malformations can be investigated. After careful dissection, the metanephric rudiments are transferred onto a filter that floats on culture medium and can be kept in a cell culture incubator for several days. However, one must be aware that the conditions are artificial and could influence the metabolism in the tissue. Also, the penetration of test substances could be limited due to the extracellular matrix and basal membrane present in the explant.
One main advantage of organ culture is that the experimenter can gain direct access to the organ. This technology is cheap, simple, and allows a large number of modifications, such as the addition of biologically active substances, the study of genetic variants, and the application of advanced imaging techniques.
Introduction
The mammalian kidney is derived from two primordial structures with mesodermal origin: the tubular epithelial ureteric bud and the metanephric mesenchyme. During nephrogenesis, the ureteric bud invades the metanephric mesenchyme and branches to form the collecting system. The metanephric mesenchyme gives rise to the epithelial elements of the nephrons. These processes occur in a precisely timed and spatially coordinated manner and are initiated by reciprocal inductive mechanisms. Both tissue components communicate and affect the other’s cell morphogenesis.
In the 1920s, it was Boyden who performed the in vivo obstruction of the mesonephric duct in chicken, providing the first indication of inductive interactions as separated nephric blastema fail to differentiate1. At about the same time, the first successful attempts to culture chicken nephric rudiments in a hanging drop were published. Subsequently, the organ culture was developed to study tissue interactions in mammalian organogenesis. In the 1950s, Grobstein developed a technique in which metanephric rudiments could be cultured on a filter. This technique was modified by Saxén, who placed the filter on a Trowell-type screen in a culture dish1. Over the years, many modifications and applications for organ culture have emerged. The method described here is based on Saxén’s technique but is simplified, as the filters float free on the medium and the diameter of the culture well only slightly exceeds the diameter of the filter, limiting unwanted movement of the filter.
Whole-organ culture is a classical, cheap, and simple but powerful tool to investigate cellular processes and intercellular communication during organogenesis. Organ culture allows for treatment with biological agents, such as growth factors, antibodies, antisense oligonucleotides, viruses, and peptides, as well as with pharmaceutical compounds and other chemicals. Also, gene function may be studied using explants derived from genetically modified mice or using inducible gene inactivation technology, such as the Cre-loxP system. This allows for the study of genetic mutations that cause embryonic lethality prior to the development of the kidney. Organ culture can also be combined with fluorescent tagging for gene function or lineage tracing and modern imaging techniques, which enable real-time monitoring of cell behavior2.
In the specific example provided here, the effect of EphrinB2-activated Eph-receptor signaling on the branching morphology of the ureteric bud was investigated. The morphology of the EphA4/EphB2 double-knockout mice suggested several severe defects in kidney development, which were detectable as early as embryonic day 11 (E11) and involved the ureteric bud, the ureter, and the common nephric duct3. Signaling via Eph receptors requires the clustering of the ligand-receptor dimer4. To over-activate Eph signaling, the kidney rudiments from E11.5 mouse embryos were cultured in the presence of clustered recombinant EphrinB2-Fc. EphrinB2 is a known ligand for the EphA4 receptor, which is expressed in the ureteric bud tips3.
Protocol
Mice were maintained according to Swedish regulations and European Union legislation (2010/63/EU). All procedures were performed following the guidelines of the Swedish Ethics Committee (permits C79/9, C248/11, and C135/14). Procedures at Heidelberg University involving animal subjects have been approved by the Regierungspräsidium Karlsruhe and the Animal Welfare Officers at the University of Heidelberg.
1. Preparation of Reagents and Materials for Culture
NOTE : Use a laminar flow hood to minimize contamination.
- On the day of dissection, pre-cluster human Fc alone or recombinant chimeric ephrin protein fused to human Fc by mixing it with anti-human Fc goat antibodies at a molar ratio of 1:5 in sterile phosphate-buffered saline (PBS). Incubate for 1 h at 37 °C5.
- Prepare the culture medium by supplementing Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12) with 1% penicillin/streptomycin (v/v), 1% glutamine (v/v), 1% insulin-transferrin-selenium (v/v), 50 µg/mL human holo-transferrin, and 1.5 µg/mL amphotericin.
NOTE: 10 mL of medium is sufficient for 16 embryos. - Add clustered recombinant ephrin solution at a final concentration of 20 µg/mL to the culture medium. Use clustered human Fc as control.
- Prepare culture plates by adding 500 µL of culture medium to each well of a sterile, uncoated, flat-bottom polystyrene 4-well plate.
- Using sterile forceps, carefully place one polycarbonate membrane filter with a pore size of 5 µm per well on top of the medium. Transfer the prepared plate to the incubator (37 °C/5% CO2). Ensure that the filter stays dry on the upper surface and floats.
NOTE: One filter is sufficient for two kidney anlagen. - Using a razor blade, cut approximately 30 pipette tips (volume: 100 µL) approximately 1 mm from the tip to result in a wider opening. Place the tips back into a pipette-tip rack.
2. Dissection of Metanephric Rudiments at E11.5
- Sacrifice a timed-pregnant mouse at E11.5 by cervical dislocation (or using a method approved by the local ethics committee).
- Remove the uterus and the embryos, as described by Brown et al 6. From this point forward, work under a dissection microscope.
- Using No. 5 watchmaker forceps, cut the embryo directly under the forelegs. Use a fresh petri dishfor each embryo. Remove the legs and the tail of the embryo. To do this, grab the tissue to be removed with one pair of No. 5 watchmaker forceps and use the tips of a second pair to cut.
- To remove the ventral organ mass, use the tips of a pair of forceps to laterally open the body wall of the embryo in a rostral-to-caudal direction.
- Cut superficially, parallel to the dorsal aorta. Use the blood-filled and therefore visible dorsal aorta for orientation. Carefully separate the ventral part of the body wall from the dorsal part using the tips of closed forceps and flip the embryo over. Cut the body wall laterally in a rostral-to-caudal direction and using the dorsal aorta for orientation, as for the previous side.
- Once the ventral body wall has been separated from the dorsal body, grab it with one pair of forceps and carefully pull to remove it, together with the organ mass.
NOTE: The metanephric anlagen usually stay attached to the dorsal body wall. They are oval, ~ 200 µm long structures located at the level of the hind limbs. - Hold the dorsal body wall with one pair of forceps, with the ventral side facing up. Using the other pair of forceps, grab the dorsal aorta at its rostral end and carefully peel the dorsal aorta from the dorsal body wall.
NOTE: Both metanephric kidneys usually stay attached to the dorsal aorta. - Using one pair of forceps, cut rostrally to the kidney anlagen to remove the aorta. Then, carefully separate the two kidney anlagen using the tips of the forceps.
- Carefully peel off unwanted tissue from the kidney anlagen using the tips of the forceps. Take care not to damage the mesenchyme, as damage can result in poor growth and exclusion of the explant from further analysis.
- Transfer the kidney anlagen separately using a microliter pipette and a wide-open pipette tip in 10 µL of PBS to the polycarbonate membrane filter in a 4-well-plate.
NOTE: Due to surface tension, the explant will be covered with a thin layer of medium. One filter is sufficient for two kidney anlagen.- Place each of the two kidney anlagen at the center of each respective half of the filter. Transfer the plate back to the incubator at 37 °C/5% CO2. Leave the plate in the incubator until the metanephric rudiments from the next embryo are ready to be placed on the filter.
- Repeat steps 2.3-2.9.1 for each embryo.
NOTE: With some practice, the dissection procedure will take about 5 min per embryo. - Incubate the kidney anlagen for 3 days at 37 °C/5% CO2.
3. Preparation of Reagents for Fixation and Staining
- To prepare 4% paraformaldehyde (PFA) in PBS without calcium and magnesium (w/v), dissolve the PFA at 60 °C in PBS, continuously stirring. Cool to room temperature and use a paper filter to remove the remaining particles. Aliquot and keep at 4 °C for immediate use or freeze for long-term storage.
CAUTION: PFA is toxic. Wear the personal protective equipment specified in the local safety standards and work in a fume hood. Discard waste following institutional guidelines and using designated waste containers. - To prepare permeabilization solution, dilute Triton X-100 in PBS without calcium and magnesium to 0.3% (v/v).
- To prepare blocking solution, add 5% (v/v) goat serum and 0.1% Triton X-100 to PBS without calcium and magnesium. Add sodium azide to a final concentration of 0.02% (w/v). Store at 4 °C.
CAUTION: Sodium azide is toxic. Handle sodium azide in accordance with the local safety and environmental protection regulations.
NOTE: When using antibodies for staining, use 5% serum from the species the secondary antibody was derived from to make blocking solution. - Dilute biotinylated Dolichorus biflorus agglutinin 1:200 in blocking solution. Dilute Alexa488-conjugated streptavidin 1:200 in blocking solution.
- To make ready-to-use embedding medium (see the table of materials), add 0.1 g of water-soluble polyvinyl alcohol-based mucoadhesive/mL to 25% (w/v) glycerol in 0.1 M Tris-HCl (pH 8.5). Heat to 50 °C under continuous agitation for 1 h, cool down, and adjust the pH to 8.0-8.5 using 1 M NaOH. Avoid higher NaOH concentrations. Add 100 µg/mL N-propylgallate and thimerosal to a final concentration of 0.02% (w/v). Stir for 30 min at room temperature.
CAUTION: Thimerosal is toxic. Handle it in accordance with the local safety and environmental protection regulations.- Fill the embedding medium in 50 mL tubes, balance the rotor, and centrifuge at 3,200 x g and room temperature for 10 min. Decant the clear solution into 15 mL tubes and discard the pellet. Aliquot and store at -20 °C for several weeks. Once thawed, keep the solution at 4 °C. Warm to ~30 °C immediately before use.
- Prepare as many glass slides as filters with explants are to be mounted. For this, glue two small coverslips, 18 x 18 mm, on either side of the glass slide using instant adhesive, Leave a ~15 mm space for the filter in between.
4. Fixation and staining
- After a culturing period of 3 days in vitro (div), remove the plates from the incubator and carefully aspirate the culture medium from each well using a micropipette. Make sure not to touch the filter and keep the tip opening towards the wall of the well.
- Add 500 µL of 4% PFA/PBS to each well. The filters will float on the fixative. Using a micropipette, carefully add the PFA fixative dropwise to submerge the filters. Incubate at room temperature for 30 min.
NOTE: For all following steps, care must be taken not to wash off the explants from the filter. Keep the opening of the pipette tip towards the wall of the well. - Remove the PFA and rinse twice with 500 µL of PBS, submerging the filter. Add 500 µL of permeabilization solution to each well and incubate at room temperature for 1 h.
- Rinse twice with 500 µL of PBS and add 500 µL of blocking solution to each well. Incubate at room temperature for 1 h.
- Replace the blocking solution with 250 µL of biotinylated Dolichorus biflorus agglutinin diluted 1:200 in blocking solution and incubate at 4 °C for 24 h.
- Remove the Dolichorus biflorus agglutinin solution and rinse twice with 500 µL of PBS per well.
- Add 250 µL of Alexa488-conjugated streptavidin diluted 1:200 in blocking solution and incubate at 4 °C for 24 h. Alternatively, incubate at room temperature for 2 h.
NOTE: A better signal-to-noise ratio is achieved when incubated at 4 °C. - Rinse twice with 500 µL of PBS per well. Using forceps, transfer the filters to the prepared glass slides, with the explants facing up. Mount with ~50-100 µL of embedding medium. Cover with 60 mm-long No. 1.5 coverslips. Allow the embedding medium solution to solidify for 2 h at room temperature in the dark. Proceed to imaging or store the slides, wrapped in foil, at 4 °C until ready to image.
- Image with a widefield epifluorescence microscope7 coupled to an Hg-lamp and use a mirror unit for blue excitation (excitation bandpass, 460 – 495 nm; dichromatic mirror, 505 nm; and emission bandpass, 510 – 550 nm) and 20X Plan-Apochromat lenses, 0.75 numerical aperture.
Representative Results
Metanephric kidney anlagen were derived from pregnant Black-6 inbred mice at E11.5 and were cultured. After 3 days, the ureteric bud had branched up to 5 times, resulting in a ramification of the initially T-shaped ureteric bud. Each explant was photographed, and the numbers of segments and endpoints were quantified to determine the branching generations and to calculate the number of endpoints per branch (Figure 1). ImageJ (rd generation; the 4th generation was reached in only 8% of the treated explants, compared to 35% of the control explants (Figure 1b). Accordingly, the number of endpoints per branch and endpoints per mm2 was reduced in the explants treated with clustered EphrinB2. In addition, a third of the explants had unusual morphology in the ureteric bud tips (Figure 1). These results suggest that EphrinB2 might have a restricting effect on the ureteric bud branching process, most likely by activating EphA4 and EphB2 forward signaling.
For a successful experiment, it is critical that the metanephric mesenchyme is not damaged during dissection. Any injury of the mesenchyme decreases the inductive potential, leads to reduced or absent ureteric bud branching, and could be a source of bias. The example in Figure 2A shows an explant where the mesenchyme is almost missing. The ureteric bud did not branch beyond the T-stage. Figure 2B shows an example where the damage of the metanephric mesenchyme led to poor growth and branching. Both explants must be excluded from analysis.
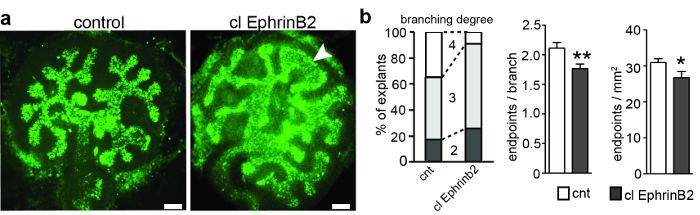
Figure 1: E11.5 Metanephric Kidney Anlagen Cultured for 3 div and Treated with Clustered Recombinant EphrinB2 or Clustered Human Fc as a Control. (A) Metanephric kidney anlagen were dissected at E11.5, cultured for 3 div, and stained with biotinylated Dolichorus biflorus agglutinin and Alexa488-conjugated streptavidin. The explants were imaged with a widefield epifluorescence microscope with an excitation bandpass of 460-495 nm, a dichromatic mirror of 505 nm, an emission bandpass of 510-550 nm, and 20X Plan-Apochromat lenses. The application of clustered recombinant EphrinB2 (clEphrinB2) resulted in reduced branching complexity and malformation of the ureteric bud tips (arrowhead). (B) The left graph shows branching generations in clEphrinB2-treated and control explants. The 4th generation of branching was reached in only 8% of the treated explants, compared with 35% of the control explants. The number of endpoints per branch (middle graph) and endpoints per area (right graph) was reduced (endpoints per branch CNT, 2.1 ± 0.09; clEphrinB2, 1.7 ± 0.08, P = 0.007 **; endpoints per square millimeter: CNT, 31 ± 0.01; clEphrinB2, 27 ± 0.02; P = 0.04*; n = 23). The data are presented as the mean ± SEM and an unpaired Student's t-test was used. Scale bar = 100 µm. This figure has been modified from Peuckert et al., 20163. Please click here to view a larger version of this figure.
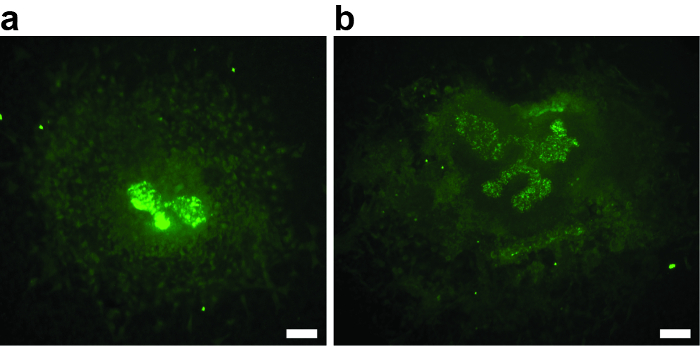
Figure 2: Examples of Two E11.5 Metanephric Kidney Anlagen that were Damaged During the Dissection, Cultured for 3 div, and Stained with Biotinylated Dolichorus Biflorus Agglutinin and Alexa488-conjugated Streptavidin. (A and B) The damage of the mesenchyme resulted in poor or absent growth and ureteric bud branching, disqualifying the explants from further analysis. (A) Metanephric kidney explant after 3 div, with absent ureteric bud branching. Only the first T-stage branch is visible. (B) Metanephric kidney explant after 3 div, with poor ureteric bud branching. Scale bar = 60 µm. Please click here to view a larger version of this figure.
Discussion
This manuscript describes a method to isolate the developing metanephric anlagen from the mouse embryo and to culture the organ rudiments. This method is a standard technique, as developed by Grobstein8 and Saxén9,10, and was adapted and modified by many others11,12. The success of the method depends mainly on the duration of the dissection, as explant survival and inductive potential decrease with prolonged dissection time. Care must also be taken not to damage the mesenchyme when cleaning the kidney rudiment from the surrounding tissue. Damage of the metanephric mesenchyme is often the reason for poor growth of the explants. However, dissection speed and fine motor skills greatly improve with practice.
The chemically defined medium in the presented protocol is commonly used to replace the serum-containing medium in primary cell and in vitro organ culture and contains a 1:1 (v/v) mixture of DMEM and Ham's F-12, supplemented with insulin-transferrin-selenium to support the growth and survival of the explants. Glucose and amino acid uptake, lipogenesis, and intracellular transport are facilitated by insulin. Selenium, a cofactor for glutathione peroxidase, functions as antioxidant. Transferrin is an iron carrier and helps to protect against oxygen radicals. In embryonic kidney culture, the addition of transferrin to the medium increases tubule differentiation and thymidine incorporation in a dose-dependent manner, with a maximum effect around 50 µg/mL13. Therefore, human-holo-transferrin is additionally supplemented into the medium, resulting in a final transferrin concentration of about 55 µg/mL. Many protocols, which use a simpler but chemically less defined composition, with Eagle's Minimal Essential Medium (MEM) or DMEM and 10% serum (i.e., fetal bovine serum, FBS) also give very satisfying results12,13,14,15. However, variations between different lots of FBS may occur. To avoid such variations and to exclude possible interference of growth factors present in the serum with Eph signaling, serum-free medium was chosen. The decision whether to use serum-free medium depends on the experimental setup and scientific question. Serum-free culture conditions would be particularly required when the ultimate goal is therapeutic application. Amphotericin B, the anti-fungal agent included in this protocol, can be omitted. The culturing period in the presented example was 3 days, but metanephric kidney rudiments can be cultured for up to 10 days15. In cultures exceeding 3 days, the medium should be changed every 48 h. The development of kidney rudiments in vitro recapitulates the in vivo sequence of pre-tubular aggregates, renal vesicles, and comma- and S-shaped bodies. After 3 days in vitro, glomerular-like structures have formed15,16. In longer cultures, the explant area increases further due to the continued branching of the ureteric bud. At about 5 days of culture, the nephrons have segregated into distal, middle, and proximal segments16.
Despite the relative ease and cost efficiency of the technique, allowing for versatile applications, some considerations should be kept in mind when planning experiments and interpreting results. Due to the extracellular matrix and basement membrane present in the cultured organs, the diffusion of exogenous agents and particles is limited17. Furthermore, the artificial culture conditions and manipulations may cause changes in the metabolism of the tissue, and cell behavior that differs from the in vivo situation15,18. Most noticeably, the explants lack blood supply, and the glomeruli are avascular; although the nephrons become segmented, zonation and the formation of a medulla and loops of Henle are missing14,19. Thus, the application spectrum of embryonic kidney culture is limited to the tubular structures, their branching morphology, and mesenchymal-epithelial interactions. Consequently, scientific questions targeting kidney function cannot be addressed.
Recent modifications of the culture method in which kidney rudiments are grown on coated glass in a low volume-enabled organotypic development even up to cortico-medullary zonation with extended loops of Henle15. It is also worth noting that, recently, a method to store and preserve living embryonic kidneys was published. This method enables the transport of embryonic kidney rudiments at E11.5 for several days and allows for them to be cultured later. This is especially of interest in collaborations20. The nature of whole-kidney rudiment culture allows a variety of methodological adaptations, including advanced imaging techniques. To avoid disturbing movements during live imaging, it is recommended to replace the floating with a fixed filter, such as a transwell inset. The presented technology has even been expanded to culture tissue blocks containing the whole urogenital tract. Using this expanded culture, ureter insertion into the bladder can be investigated21.
Declarações
The authors have nothing to disclose.
Acknowledgements
The authors thank Leif Oxburgh and Derek Adams for generously sharing their knowledge, Leif Oxburgh for the helpful comments on the manuscript, and Stefan Wölfl and Ulrike Müller for their technical support and Saskia Schmitteckert , Julia Gobbert, Sascha Weyer and Viola Mayer for help in the lab. This work was supported by Development, The Company of Biologists(to CP).
Materials
| DMEM/F-12 | Thermo Fisher Scientific | 21331020 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140148 | |
| GlutaMAX Supplement | Thermo Fisher Scientific | 35050061 | |
| DPBS, calcium, magnesium | Thermo Fisher Scientific | 14040117 | use for dissection |
| holo-Transferrin human | Sigma-Aldrich | T0665 | |
| Insulin-Transferrin-Selenium (ITS -G) (100X) | Thermo Fisher Scientific | 41400045 | |
| Paraformaldehyde | Sigma-Aldrich | 158127 | |
| Amphotericin B solution | Sigma-Aldrich | A2942 | |
| Triton X-100 | Sigma-Aldrich | X100 | |
| Sodium azide | Sigma-Aldrich | S8032 | |
| Thimerosal | Sigma-Aldrich | T5125 | |
| Propyl gallate | Sigma-Aldrich | 2370 | |
| Mowiol 4-88 | Sigma-Aldrich | 81381 | |
| Glycerol | Sigma-Aldrich | G5516 | |
| Biotinylated Dolichorus Biflorus Agglutinin | Vector Laboratories | B-1035 | |
| Alexa488 conjugated Streptavidin | Jackson Immuno Research | 016-540-084 | |
| Recombinant Mouse Ephrin-B2 Fc Chimera Protein, CF | R&D Systems | 496-EB | |
| Recombinant Human IgG1 Fc, CF | R&D Systems | 110-HG-100 | |
| Goat Anti-Human IgG Fc Antibody | R&D Systems | G-102-C | |
| Phosphate buffered saline tablets | Sigma-Aldrich | P4417 | use for fixation and immunostaining |
| Dumont #5, biologie tips, INOX, 11cm |
agnthos.se | 0208-5-PS | 2 pairs of forceps are needed |
| Iris scissors, straight, 12cm | agnthos.se | 03-320-120 | |
| Dressing Forceps, straight, delicate, 13cm |
agnthos.se | 08-032-130 | |
| Petri dishes Nunclo Delta treated | Thermo Fisher Scientific | 150679 | |
| TMTP01300 Isopore Membrane Filter, polycarbonate, Hydrophilic, 5.0 µm, 13 mm, white, plain | MerckMillipore | TMTP01300 | |
| Nunclon Multidishes 4 wells, flat bottom |
Sigma-Aldrich | D6789-1CS | |
| Microscope cover glass24x50mm thickn. No.1.5H 0.17+/-0.005mm | nordicbiolabs | 107222 | |
| Cover glasses No.1.5, 18x18mm | nordicbiolabs | 102032 | |
| Slides ~76x26x1, 1/2-w. ground plain | nordicbiolabs | 1030418 | |
| VWR Razor Blades | VWR | 55411-055 | |
| 50 mL centrifuge tubes | Sigma-Aldrich | CLS430828 | |
| 15 mL centrifuge tubes | Sigma-Aldrich | CLS430055 | |
| Whatman prepleated qualitative filter paper, Grade 113V, creped | Sigma-Aldrich | WHA1213125 | |
| Fixed stage research mircoscope | Olympus | BX61WI | |
| Black 6 inbred mice, male, C57BL/6NTac | Taconic | B6-M | |
| Black 6 inbred mice,female, C57BL/6NTac | Taconic | B6-F | |
| Greenough Stereo Microscope | Leica | Leica S6 E |
Referências
- Saxén, L. . Organogenesis of the kidney. Developmental and Cell Biology Series. 19, (1987).
- Lindström, N. O., et al. Integrated β-catenin, BMP, PTEN, and Notch signalling patterns the nephron. eLife. 4, e04000 (2015).
- Peuckert, C., et al. Multimodal Eph/Ephrin signaling controls several phases of urogenital development. Kidney Int. 90 (2), 373-388 (2016).
- Pasquale, E. B. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 6 (6), 462-475 (2005).
- Bonanomi, D., et al. Ret Is a Multifunctional Coreceptor that Integrates Diffusible- and Contact-Axon Guidance Signals. Cell. 148 (2), 568-582 (2012).
- Brown, A. C., et al. Isolation and Culture of Cells from the Nephrogenic Zone of the Embryonic Mouse Kidney. J Vis Exp. (50), e2555 (2011).
- Grobstein, C. Inductive interaction in the development of the mouse metanephros. J Exp Zool. 130, 319-340 (1955).
- Saxén, L., Toivonen, S. . Primary Embryonic Induction. , (1962).
- Saxén, L., Koskimies, O., Lahti, A., Miettinen, H., Rapola, J., Wartiovaara, J. Differentiation of kidney mesenchyme in an experimental model system. Adv Morphog. 7, 251-293 (1968).
- Dudley, A. T., Godin, R. E., Robertson, E. J. Interaction between FGF and BMP signaling pathways regulates development of metanephric mesenchyme. Genes Dev. 13, 1601-1613 (1999).
- Perälä, N., et al. Sema4C-Plexin B2 signalling modulates ureteric branching in developing kidney. Differentiation. 81 (2), 81-91 (2011).
- Thesleff, I., Ekblom, P. Role of transferrin in branching morphogenesis, growth and differentiation of the embryonic kidney. J Embryol exp Morph. 82, 147-161 (1984).
- Watanabe, T., Costantini, F. Real-time analysis of ureteric bud branching morphogenesis. Dev Biol. 271, 98-108 (2004).
- Sebinger, D. D. R., Unbekandt, M., Ganeva, V. V., Ofenbauer, A., Werner, C., Davies, J. A. A Novel, Low-Volume Method for Organ Culture of Embryonic Kidneys That Allows Development of Cortico-Medullary Anatomical Organization. PLoS One. 5 (5), e10550 (2010).
- Ekblom, P., Miettinen, A., Virtanen, I., Wahlström, T., Dawnay, A., Saxén, L. In vitro segregation of the metanephric nephron. Dev Biol. 84 (1), 88-95 (1981).
- Davies, J. A., Unbekandt, M. siRNA-mediated RNA interference in embryonic kidney organ culture. Methods Mol Biol. 886, 295-303 (2012).
- Saxén, L., Lehtonen, E. Embryonic kidney in organ culture. Differentiation. 36 (1), 2-11 (1987).
- Bard, J. B. L. The development of the mouse kidney embryogenesis writ small. Curr Opin Genet Dev. 2, 589-595 (1992).
- Davies, J. A. A method for cold storage and transport of viable embryonic kidney rudiments. Kidney Int. 70 (11), 2031-2034 (2006).
- Batourina, E., et al. Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat Genet. 32 (1), 109-115 (2002).
