Schematic representation of Thy-Noc and HU-based protocols for cell synchronization.
Figure 1 summarizes the steps required for U2OS cell synchronization and subsequent sample collection in order to verify progression through the cell cycle and to perform gene expression analyses.
Phospho-H3 and PI staining are good evaluation parameters to select synchronization methods.
Cell synchronization procedures must be assessed and optimized for each cell line due to inherent heterogeneity of cultured cells. Synchronization in mitosis is typically achieved with Thy-Noc protocol, and enrichment of mitotic cells after treatment with nocodazole can be assessed with antibodies specific for phosphorylated histone H3 (pH3), a typical mitotic marker. Identification of pH3 positive cells allows discriminating between mitotic cells and those with a 4C-DNA content not undergoing mitosis (all of which are considered a "G2" population by PI staining). In U2OS cells Thy-Noc protocol lead to a significant enrichment of a population with 4C DNA content (Figure 2A, upper left graph, blue population by PI staining), and the relative fraction of mitotic cells within this population was routinely quite pure (approximately 91% compared to 2% in asynchronous cells). Release from nocodazole results in a synchronized progression of cells to the next G1 phase, as determined by the accumulation of a population with 2C DNA content (Figure 2A, upper right graph, green population by PI staining) and the near disappearance of pH3 positive mitotic cells (Figure 2A, lower right graph). Thus, pH3 staining results confirmed the suitability of Thy-Noc protocol for mitotic synchronization of U2OS cells.
Synchronization of U2OS cells in G1/S was tested with thymidine and hydroxyurea, two widely used synchronyzers. Enrichment in different phases of the cell cycle was determined by PI staining and FACS analysis and percentage of cells summarized in a table (Figure 2B). A 24 h exposure of U2OS cells to Thy (2mM) was inefficient in arresting cells in G1/S boundary (Figure 2B), whereas treatment with HU or with a double round of thymidine (DT) resulted in a satisfactory arrest. However, only HU-treated cells recovered completely from arrest and progressed adequately through the cell cycle. By contrast, DT block induced a permanent G1 arrest in a significant fraction of the cell population (13.3% of cells in G1 at 6 h time point with DT vs. 3.1% with HU-based protocol), which negatively affected cell synchrony. Thus, exposure to HU is recommended as an appropriate G1/S synchronization method for U2OS cells.
Thy-Noc- and HU-based protocols are complementary for cell synchronization.
As shown in Figure 3A (0 h time point), treatment by Thy-Noc protocol efficiently arrested U2OS cells in M-phase entry (population with 4C DNA content, shown in blue) and treatment by HU protocol arrested cells in G1/S boundary (population with 2C DNA content, shown in green/red). Upon removal of the drugs, cells entered into and progressed through the cell cycle in a synchronized fashion. Cells recovered from Thy-Noc treatment were first observed in early G1 phase (population in green) and subsequently transitioned through G1 and up to S phase in a uniform fashion. Cells recovered from HU treatment progressed synchronously through S (population in red) and G2/M. Progression through the next cell cycle was concomitant to the loss of synchronicity. Consequently, time points over 15 hours were not included in these analyses because of loss of synchronicity after this time point.
Western blot analysis of Cyclin E1 (a G1/S cyclin) and Cyclin B1 (a G2/M cyclin), two proteins whose levels are tightly regulated in the cell cycle, and of the mitotic marker pH3, confirmed the cell cycle phase of each time point analyzed by FACS (Figure 3B). As expected, cells in M phase (corresponding to 0 h time point of Thy-Noc-synchronized cells) and cells in G2 to M phase (9 h to 12 h time point of HU-synchronyzed cells) showed a dramatic accumulation of cyclin B1 and undetectable levels of cyclin E1. Furthermore, Cyclin B1 levels were low to undetectable in G1 phase (3 h upon release from nocodazole) and gradual accumulation was observed as cells progressed through S (after Thy-Noc release) and into G2 (after HU release). Strong pH3 labeling of cells at 0 h upon Thy-Noc synchronization confirmed the enrichment of cells arrested at mitosis at this time point. Slight increase on pH3 levels at 12 h after release from HU revealed that the majority of cells with 4C DNA content were still in G2 while the proportion of cells in mitosis was increased at 15 h time point. By contrast, the expression pattern of Cyclin E1 showed a progressive accumulation from early G1 to S-phase entry (Thy-Noc procedure) and gradual disappearance in G2/M phase (HU procedure).
Expression of genes that are differentially regulated in the cell cycle can be accurately analyzed with a combination of Thy-Noc- and HU-based synchronization.
As a proof-of-concept that cell cycle-regulated gene expression analysis is best assayed by a combination of two cell synchronization methods, mRNA expression of two E2F family members known to have different expression kinetics in the cell cycle (E2F1 and E2F7) were examined by reverse transcriptase quantitative PCR (RT-qPCR). To this end, Thy-Noc and HU synchronization protocols were used, as summarized in Figure 1, and cell cycle distribution was monitored by flow cytometry as shown in Figure 3A. Figure 4 shows E2F1 (upper two graphs) and E2F7 (lower two graphs) mRNA profiles through the cell cycle. The transcriptional regulation profile of E2F1 encoding gene was best observed with Thy-Noc procedure, whereby E2F1 expression started to gradually increase from early G1 phase to reach a peak at late G1 phase (9 h after Thy-Noc release; green background). Its levels decreased thereafter, concomitant with an entry into S and G2 phases (red and blue backgrounds). By contrast, E2F7 gene expression kinetics were best detected after HU-based synchronization (lower graph; red background), with a peak expression at 6 h from release (corresponding to S to G2 phase transition). Thy-Noc procedure, on the other hand, was suitable for observing induction of E2F7, but not its downregulation. Of note, extension of the analysis beyond 15 h time point reproduced E2F1 and E2F7 mRNA expression profiles of earlier time points, although with a diminished range of mRNA variation (data not shown). Altogether, these results confirm the importance of working with a properly synchronized cell population to assess not only gene expression kinetics but also the accurate amplitude of changes in the mRNA levels.
Cell synchronization provides a better understanding of the gene expression program impacted by anticancer therapy.
To analyze the effect of the antitumor drug mitomycin C (MMC) in cell cycle dynamics as well as in gene expression, U2OS cells were first synchronized with HU and subsequently treated with MMC. Exposure to this genotoxic agent activated a checkpoint in G2 phase that was evident after 15 h of exposure (Figure 5A, lower row, blue peak). By contrast, untreated cells progressed normally through G1 at this time point (Figure 5A, upper row, green peak). Long-term MMC treatment (36 h) revealed a permanent arrest of cells in G2 phase while cells with no MMC treatment exhibited a cell cycle distribution that was similar to that shown by the asynchronous cell population.
E2F1 and p21Cip1 (CDKN1A) were chosen for gene expression analysis in MMC-treated cells (Figure 5B), given their different cell cycle-dependent regulation. Expression of E2F1 is G1 phase-dependent, as described in Figure 4, whereas induction of p21Cip1 expression is coupled to cell cycle arrest/checkpoint activation24. As shown in Figure 5B (upper left graph), E2F1 gene expression kinetics were similar in the presence or absence of MMC, as cells progressed through G1/S and into S phase. Differences in E2F1 gene expression among MMC treated and untreated cells were observed after 15 h of exposure to MMC, whereby MMC-treated cells expressed lower E2F1 mRNA levels than control cells (Figure 5B, compare solid and dashed lines). However, despite the clear difference in expression, it cannot be concluded that MMC downregulates E2F1 expression, because cell cycle profiles of MMC treated and untreated cells are clearly different in these later time points, owing to a sustained G2 phase arrest imposed by MMC. In fact, low E2F1 mRNA levels after the 15 h time point in MMC-exposed cells probably reflect an effect of the drug in cell cycle dynamics, rather than an effect of MMC in E2F1 transcriptional regulation.
In contrast to E2F1, p21Cip1 is an MMC-responsive gene. Elevated levels of p21Cip1 were detected at HU-imposed G1/S arrest, indicating G1 checkpoint activation by HU (Figure 5B, 0 h time point) and its expression decreased thereafter in non-MMC treated cells, consistent with an unperturbed cell cycle progression after release from arrest. By contrast, MMC treatment led to elevated p21Cip1 mRNA levels (Figure 5B, upper right graph, solid line) while cells were accumulating in G2 phase (9 h of MMC exposure). Despite the fact that cell cycle profiles were similar at the 9 h time point in control and MMC treated cells, gene expression analysis shows a fundamental difference. MMC exposed cells displayed an activated G2 checkpoint, evidenced by induction of p21Cip1 expression, whereas untreated cells were undergoing an unperturbed transition through G2, with low p21Cip1 levels.
Data obtained by flow cytometry analysis (Figure 5A) and data resulting from RT-qPCR (Figure 5B, upper graphs) were combined in a single graph for each of the selected genes (Figure 4B, lower graphs). mRNA expression levels where shown as relative height of the bars while the cell cycle distribution of cells for each sample was illustrated with the proportion of colors: green for G1 phase (2C DNA content), blue for G2 phase (4C DNA content) and red for S phase (intermediate DNA content). This combined method of graphical representation may be helpful for interpreting cell cycle distribution and gene expression analyses.
In summary, gene expression analysis coupled to a culture synchronization method allowed the identification of a genotoxic agent-responsive gene (p21Cip1) and led to propose that changes observed in E2F1 expression between cells treated and untreated with the agent were indirect, and possibly related to differences in cell cycle distribution of cells.
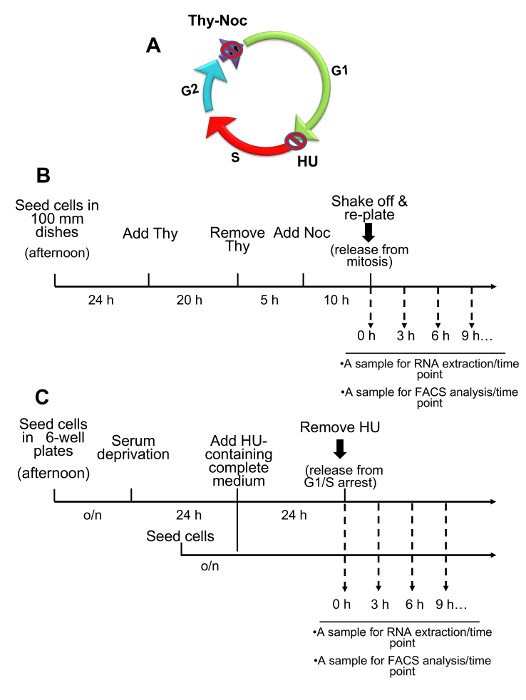
Figure 1: Overview of Two Complementary Cell Synchronization Protocols: Thymidine-Nocodazole and Hydroxyurea. (A) Graphical representation of the mammalian cell cycle indicating the points at which cell cycle arrest is achieved by cell synchronization methods. Thy-Noc-based procedure blocks cells in early mitosis (M) while HU blocks cells at the G1/S boundary. (B) Schematic representation of Thy-Noc protocol of cell synchronization. Shown are the steps typically used for gene expression analysis and cell cycle monitoring in U2OS cells. (C) Schematic representation of HU-based protocol of cell synchronization. Shown are the steps typically used for gene expression analysis and cell cycle monitoring in U2OS cells. A shorter synchronization procedure omitting serum starvation can also be applied if optimal synchronization is already achieved by a 24 h exposure to HU. Please click here to view a larger version of this figure.
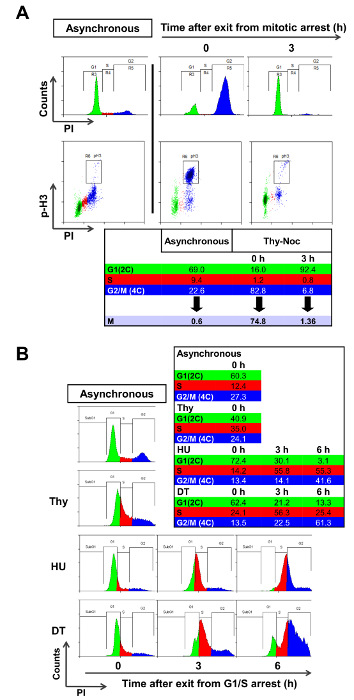
Figure 2: Assessment of Suitable Synchronization Methods. (A) Asynchronous and Thy-Noc synchronized U2OS cell cultures were subjected to pH3 staining in order to assess the fraction of mitotic cells in each case. Simultaneous PI staining allowed monitoring of the DNA content of cells. In the asynchronous population only a minimal fraction of cells with 4C DNA content (in blue) were positive for the mitosis marker. By contrast, Thy-Noc protocol efficiently accumulated cells in mitosis (positive for the pH3 staining) and allowed proper progression to the next G1 phase (in green) upon removal of nocodazole (3 h time point). Percentage of cells in each phase is summarized in the table and labeled with colors according to those employed in the PI histograms: green for 2C DNA content cells (G1), blue for cells with 4C DNA content (including G2 and M cells and named as G2 in the histograms in order to simplify the figure, and red for cells with intermediate DNA content (S). (B) Thymidine and HU were assessed for their cell cycle synchronization efficiency in U2OS cells. Cells were treated with thymidine (one or two rounds), or with HU for 24 h, and cell synchronization was examined by PI staining and FACS analysis. Summit was used for FACS data analysis. Please click here to view a larger version of this figure.
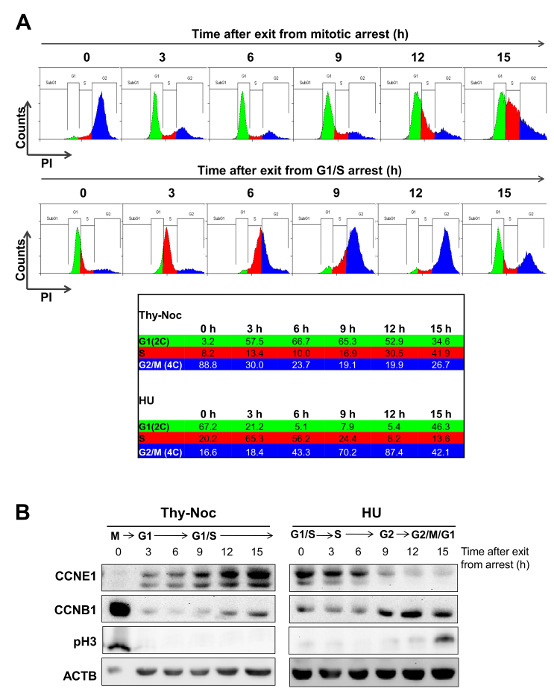
Figure 3: Outlining the Temporal Frame for Uniform Progression of Cells after Thy-Noc- and HU-mediated Synchronization. (A) U2OS cells were synchronized in M phase by Thy-Noc protocol (upper row) or in G1/S transition by HU protocol (lower row). Cell cycle progression was monitored by PI staining and FACS analysis of the DNA content of cells every 3 h after the release from chemicals. Summit was used for FACS data analysis. Cells released from Thy-Noc-mediated arrest entered synchronously in early G1 phase after 3 h, and progressed uniformly up to S phase at subsequent time points. Cells released from HU-mediated arrest transitioned synchronously through S and G2 phases. Cells collected 15 h after release represented the limits for cell synchronization, as progression to next phase was only achieved by a fraction of the population. (B) Cell synchronization was monitored by Western blot analysis by collecting protein samples every 3 h and up to 15 h following release. Expression kinetics of Cyclin E1 (CCNE1), a cyclin predominantly accumulated through the S phase, cyclin B1 (CCNB1), mainly present from G2 to M phase and pH3, a marker for mitotic cells, corroborated cell cycle distribution profiles observed by flow cytometry. Actin B (ACTB) was used as an endogenous loading control because its levels are not cell cycle-dependent. (As modified from Mitxelena et al.8). Please click here to view a larger version of this figure.
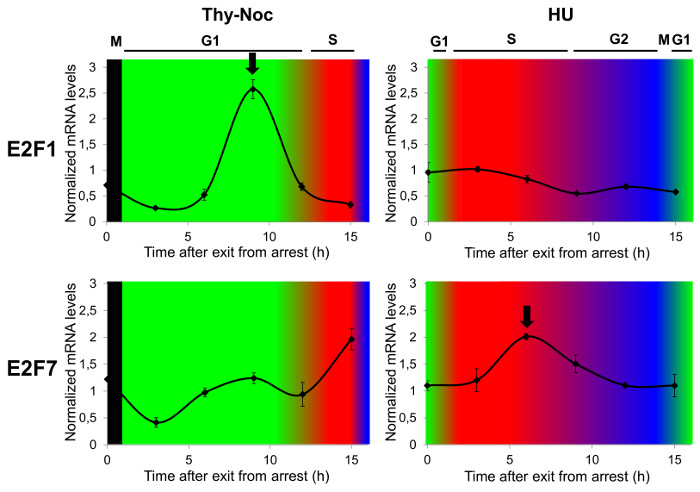
Figure 4: Study of Cell Cycle Phase-dependent Transcription of E2F family Members by Complementary Cell Synchronization Methods. U2OS cells were arrested in G2/M by Thy-Noc protocol or in G1/S by HU treatment, and samples were collected for RNA extraction and FACS analysis every 3 h up to 15 h upon release from arrest. Gene expression analysis by RT-qPCR revealed an E2F1 expression profile circumscribed mainly to G1 (upper left graph) while E2F7 gene expression profile was shifted to S phase, with a gradual downregulation through G2 phase (lower right graph). TBP was used as endogenous non-cell cycle-regulated gene to calculate relative changes in mRNA levels and results were represented as the mean values and standard deviation (SD). Progression through cell cycle was determined by PI staining and FACS analysis of the DNA content of cells. Cell cycle phases were represented as background colors in graphs: black (arrest at early mitosis), green (G1 phase), red (S phase) and blue (G2 phase). Please click here to view a larger version of this figure.
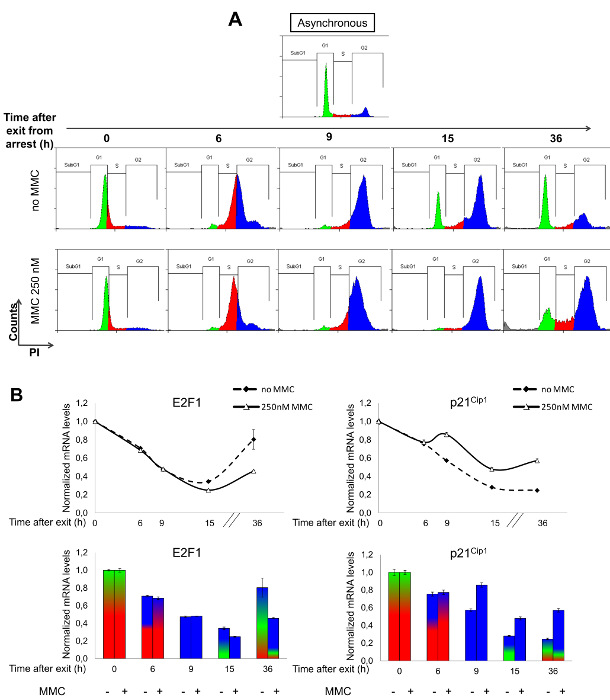
Figure 5: Impact of MMC on Gene Expression and Cell Cycle Progression. U2OS cells were treated with HU for 24 h, and MMC (250 nM) was added immediately after release from HU-mediated G1/S arrest. Samples for FACS analysis and gene expression analysis were collected at indicated time points. (A) Progression through the cell cycle was determined by PI staining and FACS analysis of DNA content by Summit software. MMC induced a moderate delay in the progression through S phase (red population) and a permanent arrest in G2 phase (blue population), as shown by the absence of cells in G1 (green population) after 15 h of treatment (lower row) compared to non-treated (upper row) cells. (B) E2F1 and p21Cip1 gene expression analysis in MMC treated or untreated cells was performed at the mRNA level by RT-qPCR. Oxa1L was used as an endogenous non-MMC-responsive gene and results were represented as mean values and SD. Upper graphs represent relative mRNA levels of selected genes after release from HU and addition of MMC. Lower graphs combine data obtained by FACS analysis and RT-qPCR. Please click here to view a larger version of this figure.
