Recombinant PFV IN is often contaminated with a bacterial nuclease1. Biochemical integration assays depend on the quantitation of the conversion of supercoiled plasmid DNA to relaxed circles and linear products. The presence of a contaminating nuclease could lead to spurious quantitation of these assays. Expression of PFV IN with a hexahistidine tag is induced in E. coli (Figure 1) and first purified by nickel affinity chromatography (Figure 2). The fractions with nearly pure PFV IN are combined and the hexahistidine tag is cleaved by a protease. We have determined that PFV IN and the contaminating nuclease have different affinities for heparin sepharose chromatography (Figure 3)1. PFV IN fractions following heparin sepharose chromatography are incubated with a supercoiled plasmid to assay for relaxed circles and linearized plasmid, the products of nuclease activity (Figure 4). This assay allows the identification of protein fractions without the nuclease (Figure 5). These fractions may be combined, dialyzed, and frozen for future experiments.
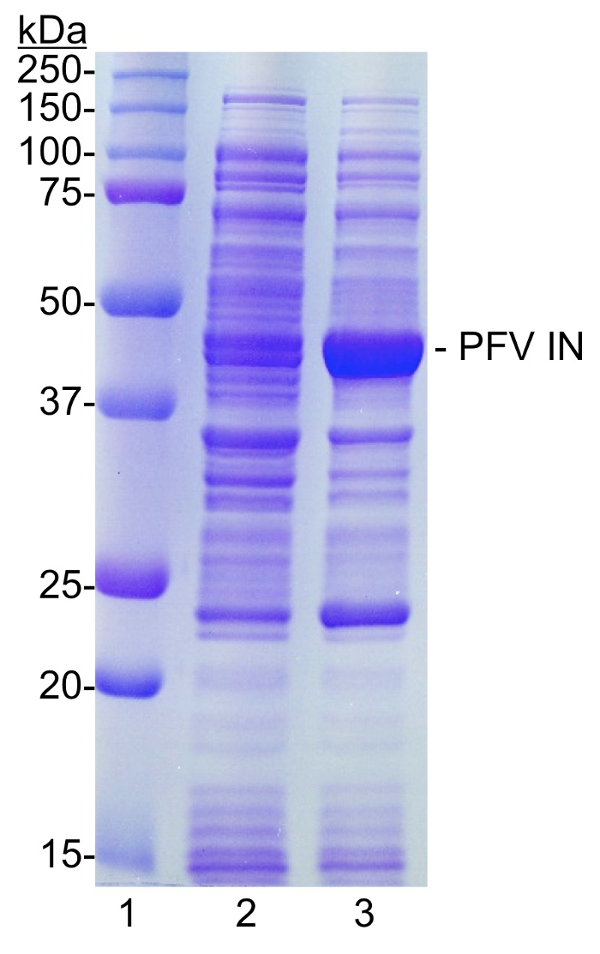
Figure 1: Induction of PFV IN in E. coli . Cells from cultures before (lane 2) and after (lane 3) induction are analyzed for the presence of PFV IN, 47374 Da. Samples were separated by 10% SDS-PAGE and stained with Coomassie brilliant blue. Lane 1, molecular weight markers (kDa). Please click here to view a larger version of this figure.

Figure 2: Nickel affinity chromatography fractions. Soluble bacterial lysates were separated by nickel affinity chromatography. PFV IN with a hexahistidine tag was eluted with a linear gradient of 20 mM to 200 mM imidazole. Fractions were evaluated by 10% SDS-PAGE and stained with Coomassie brilliant blue. PFV IN, 47374 Da, is readily apparent in the sample loaded to the column (load), but is less apparent in the flow through or wash. Fractions that elute early in the imidazole gradient commonly display a slower mobility contaminant near 75 kDa as well as some faster mobility contaminants at or below 25 kDa. At higher concentrations of imidazole there are no readily apparent contaminants and PFV IN appears to be relatively pure. In this example, fractions #33 through #84 were combined and further purified. Molecular weight markers (kDa) are on the left of each gel. Please click here to view a larger version of this figure.

Figure 3: Heparin sepharose chromatography of PFV IN 1 . Following nickel affinity and protease removal of the hexahistidine tag, PFV IN (44394 Da) was fractionated by heparin sepharose. Proteins were eluted with a linear gradient from 200 mM to 1 M NaCl. Even fractions #12 to #44 were evaluated by 8% SDS-PAGE stained with Coomassie blue. Molecular weight markers (kDa) are on the left of each gel. These fractions were tested for nuclease activity. Please click here to view a larger version of this figure.

Figure 4: Nuclease assay of heparin sepharose fractions1 . Each heparin sepharose fraction was added to supercoiled plasmid DNA. Fraction numbers correlate to those shown in Figure 3. Following incubation, the nuclease reactions were deproteinated and separated by 1% agarose with ethidium bromide. Negative controls include plasmid DNA with no protein (Target Only) and a previous purification of wild type PFV IN known to be free of nuclease (PFV IN WT). A positive control for contaminating nuclease activity is a previous purification of catalytically inactive PFV IN that is known to have contaminating nuclease (PFV IN D128N). DNA size markers are shown in kb. Supercoiled plasmid (SC), linear plasmid (L), and relaxed circles (RC) are indicated. Please click here to view a larger version of this figure.
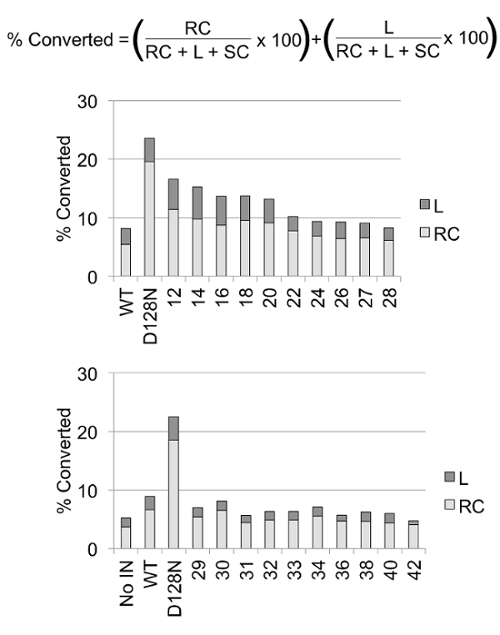
Figure 5: Quantitation of nuclease assays1. Agarose gels were scanned and pixel values were quantified for supercoiled (SC), linear (L), and relaxed circle (RC) bands in each lane. Fraction numbers correlate to those shown in Figures 3 and 4. The percentages of linear and relaxed circles were calculated using the pixel values and the equation shown. For example, the pixel values for the bands of fraction #12 are: 817636 supercoiled plasmid, 50467 linear plasmid, and 112052 relaxed circles. The total DNA pixel value for fraction #12 is the sum of these values, 980155. The percentage of linear DNA is 50467 multiplied by 100 for the percentage and divided by the total DNA value equaling 5.1%. The percentage of relaxed circles is 112052 multiplied by 100 and divided by the total DNA value equaling 11.4%. The sum of linear and relaxed circle percentages is 16.5%. Negative control plasmid DNA with no protein (No IN, bottom graph) indicates that this preparation of plasmid included 4.4% of the total DNA as linear or relaxed circles. A second negative control was a previous purification of wild type PFV IN (WT) that displayed 8.6% of the total DNA as linear or relaxed circles. These two control lanes indicate the background level of nicked or linearized plasmid present with the substrate alone and with PFV IN. A positive control is a previous purification of catalytically inactive PFV IN (D128N) which had 23% of the total DNA as linear and relaxed circles. The DNA exposed to heparin sepharose fractions #12 through #22 resulted in >10% of the DNA as linear and relaxed circles. In this example, fractions #24 to #42 displayed <10% conversion to linear and relaxed circles. These fractions were combined and dialyzed. Aliquots were frozen and stored at -80 °C for future use. Please click here to view a larger version of this figure.
