Using Phylogenetic Analysis to Investigate Eukaryotic Gene Origin
Summary
A method of constructing a phylogenetic tree based on sequence homology of SWEETs from eukaryotes and SemiSWEETs from prokaryotes is described. Phylogenetic analysis is a useful tool for explaining the evolutionary relatedness between homologous proteins or genes from different organism groups.
Abstract
Phylogenetic analysis uses nucleotide or amino acid sequences or other parameters, such as domain sequences and three-dimensional structure, to construct a tree to show the evolutionary relationship among different taxa (classification units) at the molecular level. Phylogenetic analysis can also be used to investigate domain relationships within an individual taxon, particularly for organisms that have undergone substantial change in morphology and physiology, but for which researchers lack fossil evidence due to the organisms' long evolutionary history or scarcity of fossilization.
In this text, a detailed protocol is described for using the phylogenetic method, including amino acid sequence alignment using Clustal Omega, and subsequent phylogenetic tree construction using both Maximum Likelihood (ML) of Molecular Evolutionary Genetics Analysis (MEGA) and Bayesian Inference via MrBayes. To investigate the origin of eukaryotic Sugars Will Eventually be Exported Transporters (SWEET) genes, 228 SWEETs including 35 SWEET proteins from unicellular eukaryotes and 57 SemiSWEET proteins from prokaryotes were analyzed. Interestingly, SemiSWEETs were found in prokaryotes, but SWEETs were found in eukaryotes. Two phylogenetic trees constructed using theoretically distinct methods have consistently suggested that the first eukaryotic SWEET gene might stem from the fusion of a bacterial SemiSWEET gene and an archaeal SemiSWEET gene. It is worth noting that one should be cautious to draw a conclusion based only on phylogenetic analysis, although it is useful to explain the underlying relationship between different taxa, which is difficult or even impossible to discern through experimental means.
Introduction
DNA or RNA sequences carry genetic information for underlying phenotypes that can be analyzed through physiological and biochemical methods or observed through morphological and fossil evidence. In a sense, genetic information is more reliable than evaluating external phenotypes because the former is the basis for the latter. In evolutionary study, fossil evidence is very direct and convincing. However, many organisms, such as microorganisms, have little chance to form a fossil during long geologic ages. Therefore, molecular information such as nucleotide sequences and amino acid sequences from related extant organisms are of value for exploring evolutionary relationships1. In the present study, a simple introduction of basic phylogenetic knowledge and an easy-to-learn protocol was provided for newcomers who need to construct a phylogenetic tree on their own.
Both DNA (nucleotide) and protein (amino acid) sequences can be used to infer phylogenetic relationships between homologous genes, organelles, or even organisms2. DNA sequences are more likely to be affected by changes during evolution. In contrast, amino acid sequences are much more stable given that synonymous mutations in nucleotide sequences do not cause mutations in amino acid sequences. As a result, DNA sequences are useful for comparison of homologous genes from closely related organisms, whereas amino acid sequences are appropriate for homologous genes from distantly related organisms3.
A phylogenetic analysis begins with the alignment of amino acid or nucleotide sequences4 retrieved from an annotated genome sequencing database5 listed in FASTA format, i.e., putative or expressed protein sequences, RNA sequences, or DNA sequences. It is worth noting that it is critical to collect high-quality sequences for the analysis, and only homologous sequences can be used to analyze phylogenetic relationships. Many different platforms such as Clustal W, Clustal X, Muscle, T-coffee, MAFFT, can be used for sequence alignment. The most widely used is Clustal Omega6,7 (http://www.ebi.ac.uk/Tools/msa/clustalo/), which can be used online or can be downloaded free of charge. The alignment tool has many parameters that the user can adjust before starting the alignment, but the default parameters work well in most cases. After the process is complete, the aligned sequences should be saved in the correct format for the next step. They should then be edited or trimmed using an editing software, such as BioEdit, because phylogenetic tree construction by MEGA requires the sequences to be of equal length (including both amino acid abbreviations and hyphens. In the aligned sequence, any position without an amino acid or nucleotide is represented by a hyphen "-"). Generally, all of the protruding amino acids or nucleotides at either end of the alignment should be removed. In addition, columns containing poorly aligned sequences in the alignment can be deleted because they convey little valuable information, and can sometimes give confusing or false information3. The columns containing one or more hyphens can be deleted at this time or in the later tree construction stage. Alternatively, they can be used for phylogenetic computation. When the sequence alignment and trimming is finished, the aligned sequences should be saved in FASTA format, or the desired format, for later use.
Many software platforms provide tree construction functions using different methods or algorithms. In general, the methods can be classified as either distance matrix methods or discrete data methods. Distance matrix methods are simple and fast to compute, while discrete data methods are complicated and time-consuming. For very closely related taxa with a high degree of sharing of amino acid or nucleotide sequence identity, a distance matrix method (Neighbor Joining: NJ; Unweighted Pair Group Method with Arithmetic mean: UPGMA) is appropriate; for distantly related taxa, a discrete data method (Maximum Likelihood: ML; Maximum Parsimony: MP; Bayesian Inference) is optimal3,8. In this study, the ML methods in MEGA (6.0.6) and Bayesian Inference (MrBayes 3.2) were applied to construct phylogenetic trees9. Ideally, when the proper model and parameters are used, the results derived from different methods may be consistent, and they are thus more reliable and convincing.
For a ML phylogenetic tree constructed using MEGA10, the aligned sequence file in FASTA format must be uploaded into the program. The first step then is to select the optimal substitution model for the uploaded data. All available substitution models are compared based on the uploaded sequences, and their final scores will be shown in a results table. Select the model with the smallest Bayesian Information Criterion (BIC) score (listed first in the table), set ML parameters according to the recommended model, and start the computation. The computation time varies from several minutes to several days, depending on the complexity of the loaded data (length of the sequences and number of taxa) and the performance of the computer on which the programs are run. When the computation is finished, a phylogenetic tree will be shown in a new window. Save the file as "FileName.mat". After setting parameters to specify the appearance of the tree, save once more. Using this method, MEGA can generate publication grade phylogenetic tree figures.
For tree construction with MrBayes11, the first step is to transform the aligned sequence, which is usually listed in FASTA format, into nexus format (.nex as the file type). Transforming FASTA files into nexus format can be processed in MEGA. Next, the aligned sequence in nexus format can be uploaded into MrBayes. When the file is successfully uploaded, specify detailed parameters for the tree computation. These parameters include details such as amino acid substitution model, variation rates, chain number for Markov chain Monte Carlo (MCMC) coupling, ngen number, average standard deviation of split frequencies, and so on. After these parameters have been specified, start the computation. In the end, two tree figures in ASC II code, one showing clade credibility and the other showing branch lengths, will be displayed on the screen.
The tree result will be saved automatically as "FileName.nex.con". This tree file can be opened and edited by FigTree, and the figure displayed in FigTree can be modified further to make it more suitable for publication.
In this study, 228 SWEET proteins, including 35 SWEETs from unicellular eukaryotes and 57 SemiSWEETs from prokaryotes, were analyzed as an example. Both the SWEETs and SemiSWEETs were characterized as glucose, fructose, or sucrose transporters across membranes12,13. Phylogenetic analysis suggests that the two MtN3/saliva domains containing SWEETs might be derived from an evolutionary fusion of a bacterial SemiSWEET and of an archaeon14.
Protocol
1. Sequence Alignment
- Collect amino acid sequences of eukaryotic SWEET and prokaryotic SemiSWEET in separate documents and list them in FASTA format. Download sequences from the National Center for Biotechnology Information (NCBI), European Molecular Biology Laboratory (EMBL), and the DNA Data Bank of Japan (DDBJ) databases by similarity search with the Basic Local Alignment Search Tool (BLAST) tool.
- In the example files, collect 228 putative SWEET protein sequences possessing two MtN3/saliva domains (7 transmembrane helices) of eukaryotes and 57 SemiSWEET protein sequences possessing a single MtN3/saliva domain (3 transmembrane helices) of prokaryotes13.
- To simplify the process, select 35 candidate SWEET proteins from unicellular eukaryotic organisms among the 228 putative SWEETs for phylogenetic tree construction. These sequences are attached so that the reader may practice on a real data set.
- Align the 35 SWEET sequences by inputting them into Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/).
- Copy and paste the protein sequences in FASTA format into the input box or upload a sequence file in FASTA format. Specify that they are amino acid sequence by clicking the icon under the pull-down menu in the 'STEP 1' section.
- Specify output format and other parameters in the 'STEP 2' section, if necessary. For this study, set output format as "clustal w/o number", and leave the other parameters on default settings. In most cases, the default parameters work well without any specification.
- Submit and run the alignment in the 'STEP 3' section. It may take anywhere from several seconds to minutes until the alignment is finished. In the "Result Summary" panel, right-click the link under the "Alignment in CLUSTAL format" and save the aligned sequences as "35.clustal" (Figure 1).
- Open the alignment result file in BioEdit.
- On the main panel of BioEdit, click "Sequence" and select "Edit Mood"in the first pull-down menu, then click "Edit Residues" in the sub-menu (Figure 2).
- Select the protruding sequences on the left side of the alignment with the cursor (the selected sequence will be shown in black) and click the "Delete" icon under the "Edit" menu to remove the selected sequences (Figure 3).
- Select and delete the protruding sequences on the right side of the first MtN3/saliva domain, and save the trimmed first MtN3/saliva domain sequences as 35-I.fas (Figure 4). Likewise, delete the left and right side protruding sequences of the second MtN3/saliva domain and save it as 35-II.fas. The first and the second MtN3/saliva domain sequences can be predicted with RHYTHM (http://proteinformatics.charite.de/rhythm/inndex.php?site=helix) or TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) in advance.
- Open the file 35-I.fas with MEGA, and click "align" when prompted. Under the "Edit" menu, click "Select All", then click "Select Sequence(s)"; the names and sequences of the taxa will be selected in black (Figure 5).
- Choose "Copy" from the "Edit" menu to copy the sequences onto the clipboard, and then paste the copied sequences into a doc file.
- In the doc file, replace all "#" with ">", and then delete any unrelated characters to convert them to FASTA format. Add "-I" at the end of each taxon name to mark them as the first MtN3/saliva domain sequences. Process the second MtN3/saliva domain sequence following the same method and add "-II" after each taxon name.
- Combine the first and second MtN3/saliva domain sequences in FASTA format in a doc file.
- Load the combined sequences into Clustal Omega again and align the sequences as described above. Save the result as "35 realigned.clustal".
- Open the "35 realigned.clustal" file in BioEdit, delete the uneven (protruding) amino acid residues at either end of the aligned sequences, and then save the sequences as "35 realigned.fas". Click "Yes" when warned that some non-standard characters cannot be saved.
2. Computation of the Phylogenetic Tree
- Open "35 realigned.fas" in MEGA.
- Click the "Data" menu and choose "Export Alignment", and save the alignment in PAUP format (nexus) as "35.nex" for later use in MrBayes (Figure 6).
- Meanwhile, click the "Models" icon on the main panel of MEGA, choose "Find Best DNA/Protein Models (ML)", and click "OK" on the pop-up window. Click "Compute" to begin the model searching process (Figure 7). A new progress panel will open; this process lasts several minutes to several days, depending on the complexity of the loaded sequences and the computer's performance.
NOTE: A table showing the results will open after the model searching process is finished ( Figure 8). The smallest BIC score will be listed first, followed by a series of different models with gradually increasing BIC scores. The first model "LG+G+F" with the smallest BIC score is the recommended model for ML tree based on the "35 realigned.fas" file.
- Click the "Phylogeny" icon on the main panel of MEGA, click "Construct/Test the Maximum Likelihood Tree", and then click "Yes" on the pop-up panel. A new window will open showing different parameters that need to be specified (Figure 9).
- First, set the bootstrap value in the test of the phylogeny box; 500 or 1,000 is adequate in most cases. Under the substitution model, choose "amino acid" as the substitution type. The purpose of choosing a substitution model is to estimate the true difference between sequences based on their present states3.
- Select "LG with Freqs. (+F) model" (LG+F) in the model/method box. In the rates and pattern box, select "Gamma Distributed" (G) to describe rate variations across sites, i.e., giving more weight to changes at slowly evolving sites3. In the data subset box, select "Complete deletion" to remove all of the columns containing hyphens.
- Keep all other parameters in their default states (Figure 9). After specification of these parameters, click the "Compute" icon to start the calculation.
3. Presentation of the Phylogenetic Tree
NOTE: A phylogenetic ML tree will be presented when the computation using MEGA is finished (Figure 10).
- Under the pull-down menu of the "File" icon on the tree panel, choose "Save Current Session" to save the result (.mas is the default file type). In the present study, the result was saved as "35.mas". On the tree panel, many parameters including length of clade, tree style, tree topology, font of the taxon name, size, and color, are displayed and can be set to different options.
- Save the final tree file by clicking the image icon, and save the figure in different formats or copy the image as the source for photo-editing.
4. Analysis of the Relationship of SWEETs and SemiSWEETs Using Sequence Alignment
NOTE: This step may not be needed in ordinary sequence analysis.
- Align the 228 eukaryotic SWEETs and 57 prokaryotic SemiSWEETs in Clustal Omega as described above. The alignment results can be shown in Jalview, which is integrated in Clustal Omega, and copied to save in a photo editor (Figure 11).
NOTE: In the example alignment, some SemiSWEETs from α-Proteobacteria are aligned with the first MtN3/saliva domain of the SWEET sequences, whereas SemiSWEETs from Methanobacteria (archaea) are aligned with the second MtN3/saliva domain of the SWEET sequences.
5. Phylogenetic Tree Construction with MrBayes
- For Bayesian Inferences with MrBayes, open the MrBayes executable file and a DOS interface will come up in a new window. The first step is to read the nexus data file. Input "execute 35.nex" after the prompt (remember to save the 35.nex file in the same directory of the MrBayes executable file, or point out the pathway of the file before uploading it). A "successful read matrix" message will be shown following the last of the listed taxa (Figure 12). The 35.nex file has already been prepared and saved in MEGA (see 2.1 above).
- Set the evolutionary model.
- After the prompt, type "prset aamodelpr = fixed(lg); lset rates = g". The "lg" and "g" correspond to the "LG" and "G" model which is set in MEGA. After successfully setting the model, type "mcmc nchains = 4 ngen = 5,000,000" after the prompt. Use of the "nchains=4" entry signifies a total number of one cold chain and three hot chains for Metropolis coupling. "ngen = 5,000,000" means to run 5,000,000 generations of Metropolis coupling for convergence of the cold and hot chains. In this study, average standard deviation of split frequencies below 0.01 was regarded as convergence of the hot and cold chains.
- Note that the ngen number cannot be predicted accurately at the beginning of the process, and usually needs to be adjusted based on the change in the average standard deviation of split frequencies. In addition, the ngen number for convergence may be different each time when running the program based on the same data.
- Run the analysis: This step lasts from several minutes to several days, depending on the complexity of input data and the performance of the computer. After completing the preset computation, a prompt will ask "Continue with analysis (yes/no)?" If "no" is typed after the prompt, the computing will stop (Figure 13), otherwise it will continue to compute after the number of further generations is input. When the computation is finished (with an average standard deviation of split frequencies <0.01 or 0.05), stop the computation by typing "no" after the inquiry prompt.
NOTE: 0.01 is a strict criterion, 0.05 is moderate and usually adequate. - Summarize the samples: Type "sump" after the prompt to summarize samples of model parameters (Figure 14). Then type "sumt relburnin=yes burninfrac=0.25" after the prompt to summarize tree samples. Detailed information about phylogenetic tree construction will be displayed as in Figure 15, followed by two tree figures that will appear in ASC II code on the screen, one showing clade credibility and the other showing branch lengths. At the same time, a tree file with the name of "35.nex.con" will be saved automatically.
- For a better presentation of the phylogenetic tree, open the "35.nex.con" tree file with the FigTree tool (http://tree.bio.ed.ac.uk/software/figtree/), select a style or size to display the result (Figure 16), or even edit it in a photo editor to make it more reader-friendly.
Representative Results
Phylogenetic trees show that all of the first MtN3/saliva domains of the 35 SWEET sequences clustered as one clade and the second MtN3/saliva domains of the SWEET sequences clustered as another clade. In addition, alignment results of the SWEETs and SemiSWEETs show that some SemiSWEETs from α-Proteobacteria aligned with the first MtN3/saliva domain of the SWEET sequences, whereas SemiSWEETs from Methanobacteria (archaea) aligned with the second MtN3/saliva domain of the SWEET sequences. These results together suggest that the two MtN3/saliva domains containing SWEETs might be derived from an evolutionary fusion of a bacterial SemiSWEET and of an archaeon14.
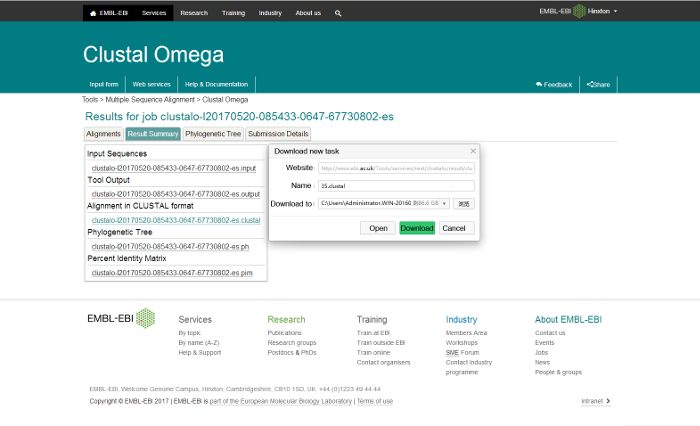
Figure 1: Save the aligned sequences of the 35 putative eukaryotic SWEETs as "35.clustal" via Clustal Omega. Please click here to view a larger version of this figure.
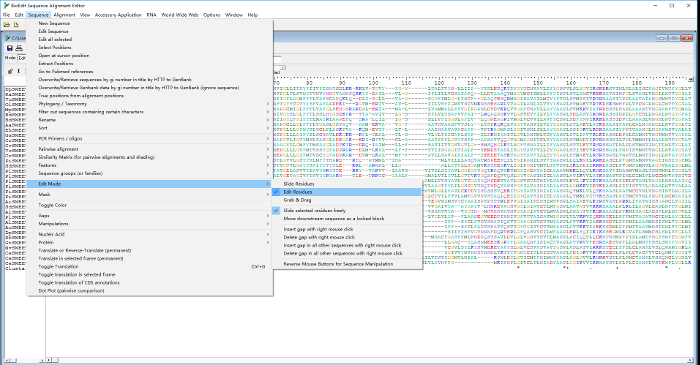
Figure 2: Select path in BioEdit to trim the aligned sequences of "35.clustal," which was prepared in Clustal Omega. Please click here to view a larger version of this figure.
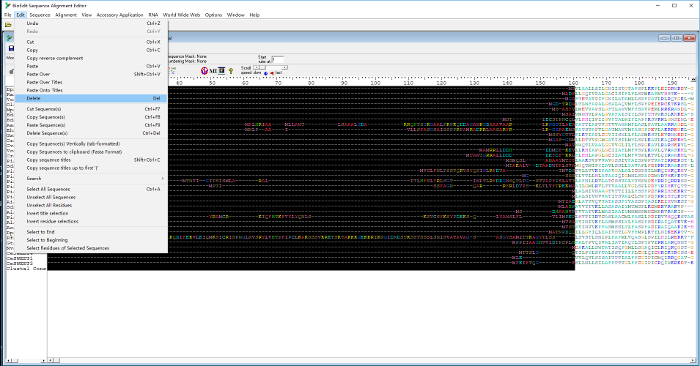
Figure 3: Select and delete the uneven sequences at the left side of the first MtN3/saliva domain sequences of the 35 putative eukaryotic SWEETs in BioEdit. Please click here to view a larger version of this figure.
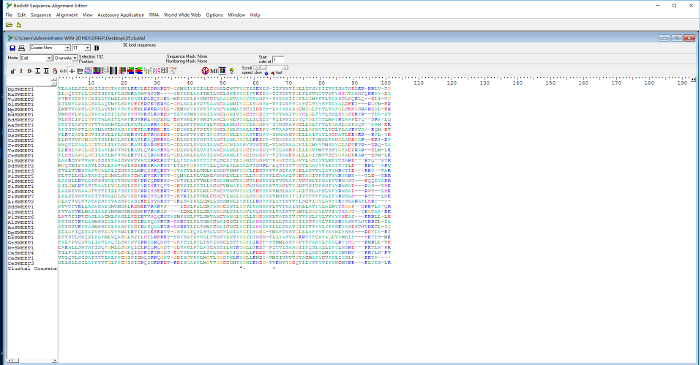
Figure 4: The trimmed sequences of the first MtN3/saliva domain of the 35 putative eukaryotic SWEETs in BioEdit. Please click here to view a larger version of this figure.

Figure 5: Select and copy the first MtN3/saliva domain sequences of the 35 putative eukaryotic SWEETs in MEGA. The copied sequences will be pasted in a doc file for the editing. Please click here to view a larger version of this figure.
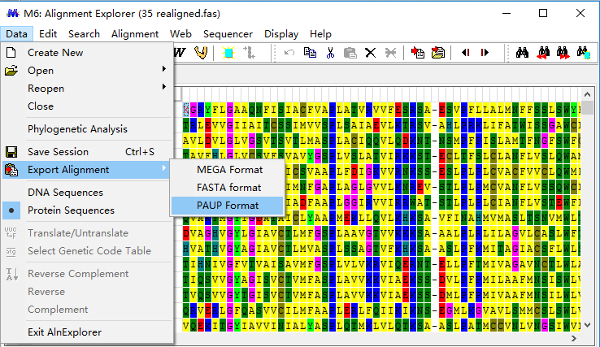
Figure 6: Convert "35 realigned.fas" into "35.nex" (PAUP format) for Bayesian Inference at a later stage. Please click here to view a larger version of this figure.
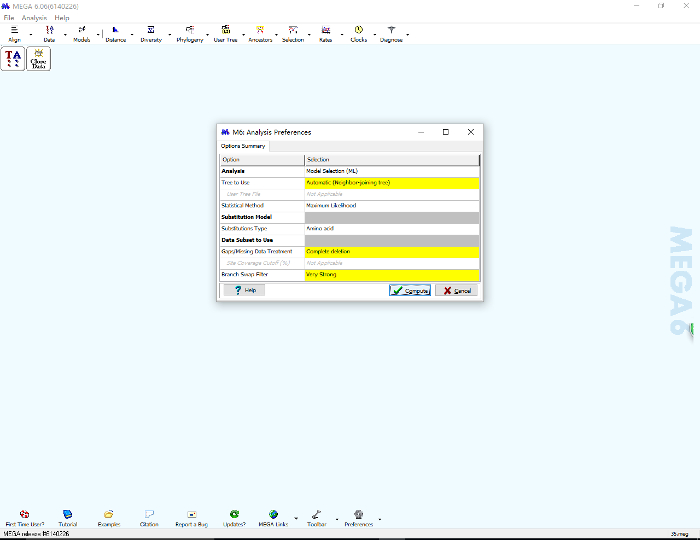
Figure 7: Search for the best-fit substitution model by MEGA for Maximum Likelihood (ML) phylogenetic tree construction based on the "35 realigned.fas" file. Please click here to view a larger version of this figure.
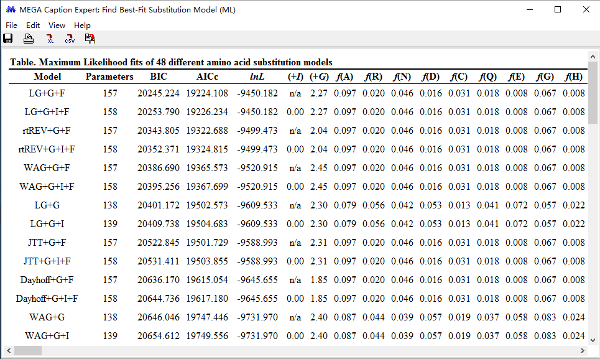
Figure 8: A table of the best-fit substitution model computed for ML tree based on the "35 realigned.fas" file. Please click here to view a larger version of this figure.
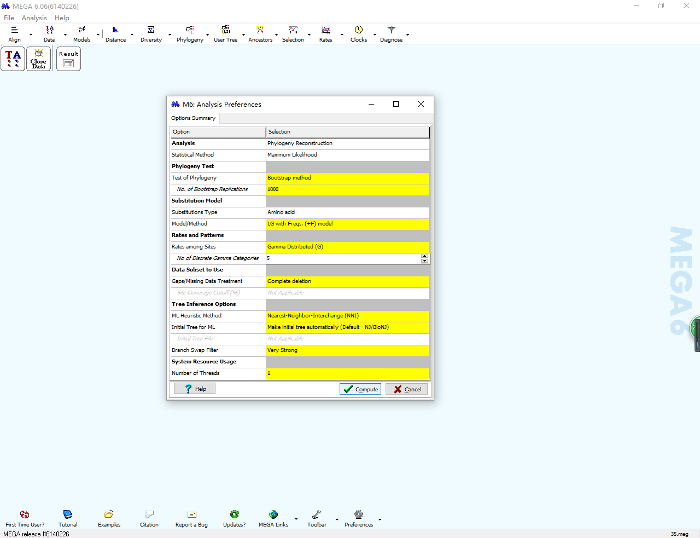
Figure 9: Specify the parameters for ML tree computation based on the best-fit substitution model for "35 realigned.fas" in MEGA. Please click here to view a larger version of this figure.
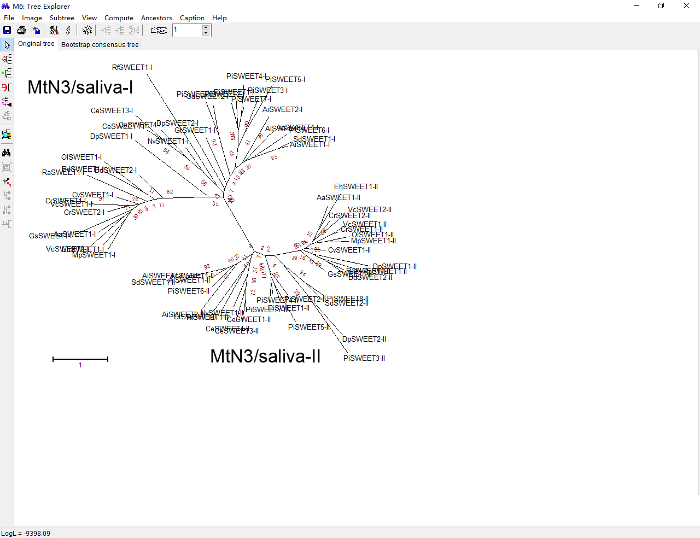
Figure 10: An original ML tree constructed by MEGA based on "35 realigned.fas". At this stage, many options for figure style, size, color, etc., are available. Please click here to view a larger version of this figure.
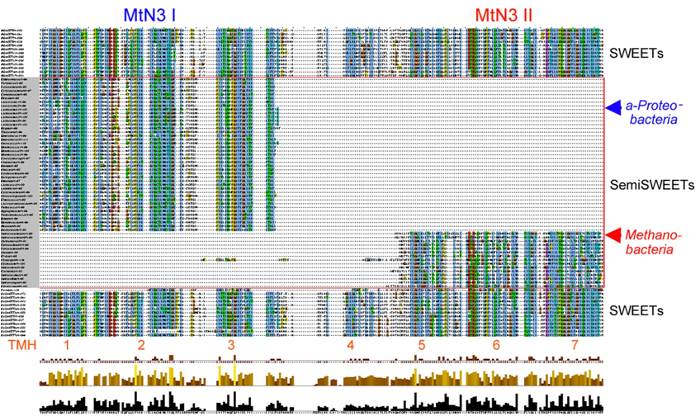
Figure 11: Alignment of 228 eukaryotic SWEETs and 57 prokaryotic SemiSWEETs by Clustal Omega. The results were shown in Jalview, integrated into Clustal Omega. In the alignment, some SemiSWEETs from α-Proteobacteria were aligned with the first MtN3/saliva domain of the SWEET sequences, whereas SemiSWEETs from Methanobacteria (archaea) were aligned with the second MtN3/saliva domain of the SWEET sequences. Please click here to view a larger version of this figure.
Figure 12: Load the "35.nex" file into MrBayes in the DOS window. In order to show the overall results, content that was similar was deleted to reduce the figure length. Please click here to view a larger version of this figure.
Figure 13: Information displayed on the screen after computation of the "35.nex" file using MrBayes. To show the overall results, content that was similar was deleted to reduce the figure length. Please click here to view a larger version of this figure.
Figure 14: Summarized samples of model parameters for the "35.nex" file. Please click here to view a larger version of this figure. Please click here to view a larger version of this figure.
Figure 15: Summarized tree samples of the "35.nex" file. To show the overall results, content that was similar was deleted to reduce the figure length. Please click here to view a larger version of this figure.
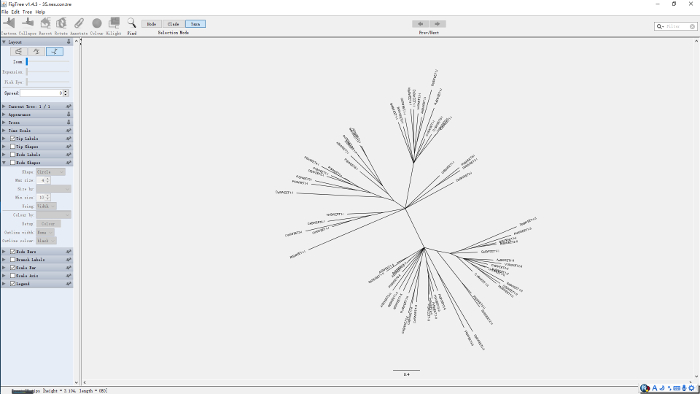
Figure 16: The phylogenetic tree of "35.nex.con" displayed by FigTree. Please click here to view a larger version of this figure.
Discussion
It is becoming increasingly popular in biological research to make a phylogenetic tree based on nucleotide or amino acid sequences8. Generally, there are three critical stages of the practice including sequence alignment, evaluation of the aligned sequences with the proper method or algorithm, and visualization of the computational result as a phylogenetic tree. In the presented study, three rounds of sequence alignment were conducted: first, the SWEET protein sequences, including the first and second MtN3/saliva domain, were aligned; second, each of the individual MtN3/saliva domain sequences of the SWEETs as an independent taxon were gathered and aligned together; and finally, SemiSWEET sequences and SWEET sequences were jointly aligned. Only one round of sequence alignment is usually needed for phylogenic tree construction.
In the preliminary stage, homologous sequences can be downloaded from NCBI or other databases. These downloaded sequences may need to be screened if they are not well annotated. In the first and second stage, alignment and computation cannot be started if the sequence format is incorrect. For example, Clustal Omega will reject any departure from the FASTA format in the sequence file. In the computational stage, note that the sequence lengths including both amino acids or nucleotides and hyphens are required to be equal before being evaluated by MEGA.
Despite the wealth of methods and models for tree construction that are available, none of them is foolproof. Robust and convincing results are those that are consistent with each other when different algorithms or models are used to evaluate the same data15. In the ML method, the reliability of tree topology largely depends on the bootstrap value of each clade; a bootstrap value of 70 or greater is generally regarded as reliable. In the present study, all of the first MtN3/saliva domain sequences clustered as a large clade with a bootstrap value of 83. The value of the other clade containing all the second MtN3/saliva domain sequences, however, was only 6 (Figure 10). To verify the tree architecture, MrBayes, which employs a completely different method16 than ML, was used to analyze the relationship of the taxa. The posterior probabilities16 of the first and second domain clades obtained from MrBayes were 100 and 68, respectively (Figure 16).
Another limitation of the ML and the MrBayes computation is that both are time-consuming to run. Using a computer with multi-core processors and graphical processing units (GPU) is helpful to improve computational performance and speed17,18. For the operation of MrBayes, a computer with a discrete graphics card and the appropriate CUDA drivers can significantly speed up the likelihood calculations11.
Selecting the proper model for phylogenetic tree computation is difficult for those with little experience. In this respect, MEGA provides an easy way to find the best model by comparing the BIC scores of candidate models. In addition, the recently upgraded MEGA 6.0 integrates several sequence alignment tools such as MUSCLE and Clustal W10, which are very convenient to use. It also provides both a sequence editing and phylogenetic tree construction function. These features partly explain why this software is so popular in the computational molecular evolution field. As for MrBayes, a significant advantage of this tool is that it can process mixed datatypes together (e.g., morphological and molecular data)11, and thus the results are more comprehensive.
In conclusion, the present study provides a method to analyze the molecular origin of protein-encoding genes that have undergone complex variation such as fusion after duplication or horizontal gene transfer (HGT) during evolution. Hopefully, more findings will be revealed with broad application of phylogenetic analysis in the evolutionary research field.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31371596), the Bio-technology Research Center, China Three Gorges University (2016KBC04), and the Natural Science Foundation of Jiangsu Province, China (BK20151424).
Materials
| Adobe Illustration | a graphical tool developed by Adobe Systems Software Ireland Ltd. Copyright © 2017 | ||
| BioEdit | a biological sequence alignment editor written for Windows 95/98/NT/2000/XP/7. Copyright © Tom Hall | ||
| Clustal Omega | a package for making multiple sequence alignments of amino acid or nucleotide sequences. http://www.clustal.org/ | ||
| CorelDRAW | a graphic design software. Copyright © 2017 Corel Corporation | ||
| FigTree | a graphical viewer of phylogenetic trees designed by the University of Edinburgh | ||
| MEGA | MolecularEvolutionary Genetics Analysis version6.0 http://www.megasoftware.net/home | ||
| MrBayes | an Bayesian phylogenetic inference tool | ||
| NVIDIA | a company designs graphics processing units (GPUs) for the gaming and professional markets. Corporation Copyright © 2017 | ||
| PAUP | Phylogenetic Analysis Using Parsimony. David Swofford's program implements the maximum likelihood method under a number of nucleotide models. | ||
| Photoshop | a raster graphics editor developed and published by Adobe Systems Software Ireland Ltd. Copyright © 2017 | ||
| RHYTHM | a knowledge based prediction of hekix contacts. Charité Berlin – Protein Formatics Group – Copyright 2007-2009 | ||
| TMHMM | a tool for prediction of transmembrane helices in proteins. http://www.cbs.dtu.dk/services/TMHMM/ | ||
| Compter | 4GB memory, Core 2 or above CPU. Windows 7, Windows 10 |
Referências
- Nei, M., Kumar, S. . Molecular Evolution and Phylogenetics. , (2000).
- Foth, B. J. Phylogenetic analysis to uncover organellar origins of nuclear-encoded genes. Methods Mol Biol. 390, 467-488 (2007).
- Baldauf, S. L. Phylogeny for the faint of heart: a tutorial. Trends Genet. 19, 345-351 (2003).
- Feng, D. F., Doolittle, R. F. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol. 25, 351-360 (1987).
- Persson, B. Bioinformatics in protein analysis. EXS. 88, 215-231 (2000).
- Sievers, F., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 7, 539 (2011).
- Sievers, F., Higgins, D. G. Clustal omega. Curr Protoc Bioinformatics. 48, 1-16 (2014).
- Yang, Z., Rannala, B. Molecular phylogenetics: principles and practice. Nat Rev Genet. 13, 303-314 (2012).
- Hall, B. G. Comparison of the accuracies of several phylogenetic methods using protein and DNA sequences. Mol Biol Evol. 22, 792-802 (2005).
- Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 30, 2725-2729 (2013).
- Ronquist, F., et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61, 539-542 (2012).
- Chen, L. Q., et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature. 468, 527-532 (2010).
- Xuan, Y., et al. Functional role of oligomerization for bacterial and plant SWEET sugar transporter family. Proc Natl Acad Sci USA. 110, 3685-3694 (2013).
- Hu, Y., et al. Phylogenetic evidence for a fusion of archaeal and bacterial SemiSWEETs to form eukaryotic SWEETs and identification of SWEET hexose transporters in the amphibian chytrid pathogen Batrachochytrium dendrobatidis. FASEB J. 30, 3644-3654 (2016).
- Holder, M. T., Zwickl, D. J., Dessimoz, C. Evaluating the robustness of phylogenetic methods to among-site variability in substitution processes. Philos Trans R Soc Lond B Biol Sci. 363, 4013-4021 (2008).
- Alfaro, M. E., Holder, M. T. The Posterior and the Prior in Bayesian Phylogenetics. Annu Rev Ecol Evol Syst. 37, 19-42 (2006).
- Suchard, M., Rambaut, A. Many-core algorithms for statistical phylogenetics. Bioinformatics. 25, 1370-1376 (2009).
- Zierke, S., Bakos, J. FPGA acceleration of the phylogenetic likelihood function for Bayesian MCMC inference methods. BMC Bioinformatics. 11, 184 (2010).
