Generation of hepatocyte–like cells: Figure 1 describes the timeframe of the changes that occur during the differentiation of human iPSCs to hepatocyte-like cells. The culture of iPSCs on E-Cad-Fc provides approximately 2 mm diameter colonies that express the pluripotent marker OCT4 (Figure 1A-B). The morphology of the cells grown on an E-cadherin matrix will be slightly different to those cultured on other surfaces. They tend to have a flattened morphology with a greater cytoplasmic surface area (Figure 1A). Although human pluripotent stem cells can be efficiently cultured on a number of different matrices11, the culture of iPSCs on an E-cadherin matrix is believed to increase the homogeneity of the pluripotent cell population and allows enzyme-free manipulation of the cells during passage12. However, several alternative matrices, including basement membrane matrix, laminins, vitronectin, and mouse embryonic fibroblasts (MEF), can also be used to maintain human iPSCs and all should be compatible with the described procedure13. In all cases, the cell pluripotency can be confirmed by Tra-1-60 or equivalent8.
To initiate the differentiation, the iPSCs should be collected as small clusters (Figure 1A). At the beginning of the differentiation, the plated cells should cover approximately 80% of the surface of the plate. After 5 days of treatment with growth factors, the cells express proteins that are characteristically expressed in the definitive endoderm such as SOX17 and FOXA2 (Figure 1B). At this stage, over 90% of the cells typically express these markers. After 5 more days of differentiation, the endoderm converts to a hepatic fate, and in addition to FOXA2, the cells express HNF4a, which can be identified throughout the remainder of the differentiation process (Figure 1B). As the cells adopt a hepatic fate, they should cover the surface of the plate as a monolayer. After addition of HGF, the cells begin to express proteins that are found in fetal hepatocytes including AFP (Figure 1B). Lipid droplets can be observed within the cytoplasm of the cells. At the completion of differentiation, the cells adopt a cuboidal morphology with a nucleus containing prominent nucleoli and a large cytoplasm with multiple lipid droplets. A subset of the hepatocyte-like cells will be multi-nucleated. Most of the cells express an extensive repertoire of hepatocyte proteins including Albumin (Figure 1B)14.
Effective high throughput screen using hypercholesterolemia as a disease model: Hypercholesterolemia reflects excessive concentrations of Low density lipoprotein-cholesterol (LDL-C) in the serum. In familial hypercholesterolemia (FH), the increased level of serum LDL-C is due primarily to mutations in the Low-density lipoprotein receptor (LDLR) that mediates uptake of LDL-C for clearance by the hepatocytes. We have previously described the generation of iPSCs from a patient with homozygous familial hypercholesterolemia and their use in generating hepatocytes that mirrored the patient's liver disease in culture9. It was demonstrated that these cells could be used successfully as a platform for drug discovery. When a library of ~2,500 drugs was screened, it was found that cardiac glycosides had an unexpected ability to lower LDL-C in iPSC-derived hepatocytes, primary human hepatocytes, and in mice with humanized livers. Moreover, upon examining patient medical records, we found that cholesterol levels fell in patients that were prescribed a cardiac glycoside for the treatment of heart disease. These studies confirmed that iPSC-derived hepatocytes could be successfully used in screens to identify therapeutics.
In order to provide a platform that is compatible with screening, it is important that the differentiations are reproducible and that well to well variation is minimized. Figure 2 shows the results of immunostaining on each well of a 96-well plate to detect HNF4a and Albumin at day 20 of differentiation. As can be seen in the figure, the distribution of these hepatocyte proteins is similar across all wells.
The central protein component of LDL-C is APOB. Here, we have used iPSCs to screen for APOB lowering compounds to provide an example of using iPSC-derived hepatocytes for small molecule screening (Figure 3A). APOB can be easily measured in solution by ELISA. Since ELISA can be performed on large numbers of samples, it can be used as a basis for a screen. The suitability of any assay for high throughput screening is best determined by using a measurement called the Z-factor (or Z')15. This statistical parameter calculates the effectiveness of an assay to distinguish between positive and negative control samples. An assay with a Z-factor >0.5 is considered compatible with a screen15. As can be seen in Figure 3B, the Z' of the APOB assay = 0.73, which confirms the suitability of using the platform for drug discovery.
Identification of compounds that have the capacity to lower APOB levels in the culture medium of iPSC–derived hepatocytes: To determine whether the platform could be used to identify novel compounds that impact APOB levels, we screened 1,000 small molecules from the South Carolina Compound Collection (SC3) Representative Set Lite (RSL). The RSL was generated by the MUSC South Carolina Center for Therapeutic Discovery, and includes compounds that are structurally diverse and reflect the parent library.
The post-drug: pre-drug ratio of APOB was determined for each compound (Figure 3C) and identification of hits was achieved by z-score analysis (Figure 3D)16. In this case, small molecules that reduced APOB with a z-score ≤-2.0 as potential hits were considered. When screening 1,000 small molecules, the number of potential hits with a z-score of ≤-2.0 is usually relatively manageable. However, when conducting a larger screen, we would rely on a more conservative score of ≤-3.0, based on the 3-sigma rule, to increase confidence in potential targets. In this example, we identified 24 potential hits with an APOB ratio that varied from 0.86 to 0.44 (Figure 4A). A secondary assay was performed on quadruplicate samples (n = 4 biological replicates) to exclude compounds that had a negative impact on cell viability and to determine which compounds could reproducibly lower APOB in the medium. Of the original 24 candidate compounds, four were found to be reproducible (p ≤0.05) (Figure 4B-C). Upon further study, 2 of these compounds were found to negatively affect cell viability, suggesting that the observed reduction in APOB was likely to a be a consequence of cell loss. The remaining two compounds would be considered good candidates for follow-up studies.
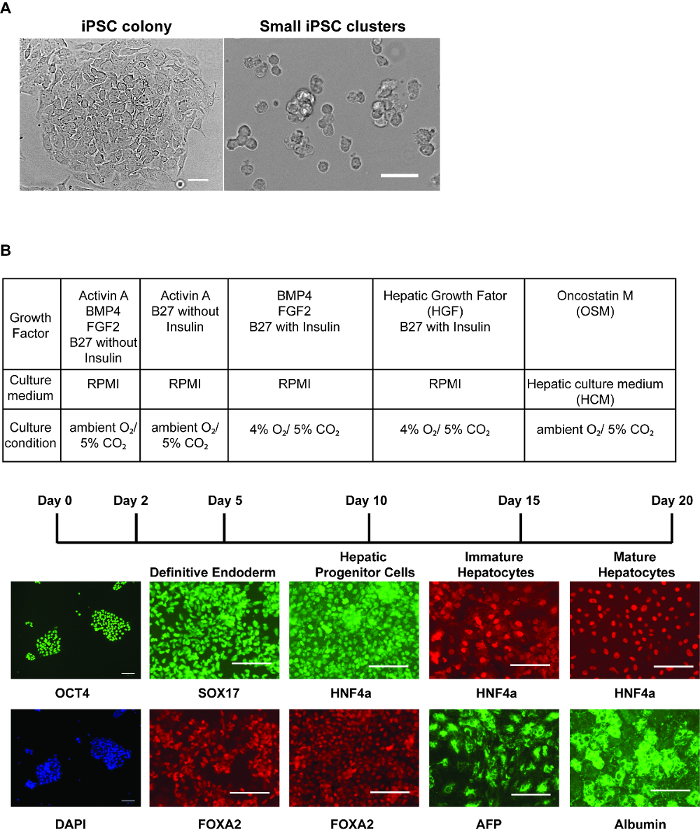
Figure 1: Step-wise hepatic differentiation of human iPSCs. (A) Morphology of iPSC colonies immediately before harvest (left panel) and iPSCs after collecting cells for differentiation (right panel) – after accutase treatment, the cells should be dissociated into 3 – 6 cells/cluster. Scale bar = 50 µm. (B) Table showing culture conditions at each stage of differentiation (upper). Immunostaining (lower panels) was performed to identify marker expression at each stage of differentiation. Panels show OCT4 and DAPI stained nuclei in iPSCs, SOX17 and FOXA2 in definitive endoderm, HNF4a and FOXA2 in hepatic progenitor cells, HNF4a and AFP in immature hepatocyte-like cells, and HNF4a and Albumin in relatively mature hepatocyte-like cells. Scale bars = 100 µm (B). Please click here to view a larger version of this figure.
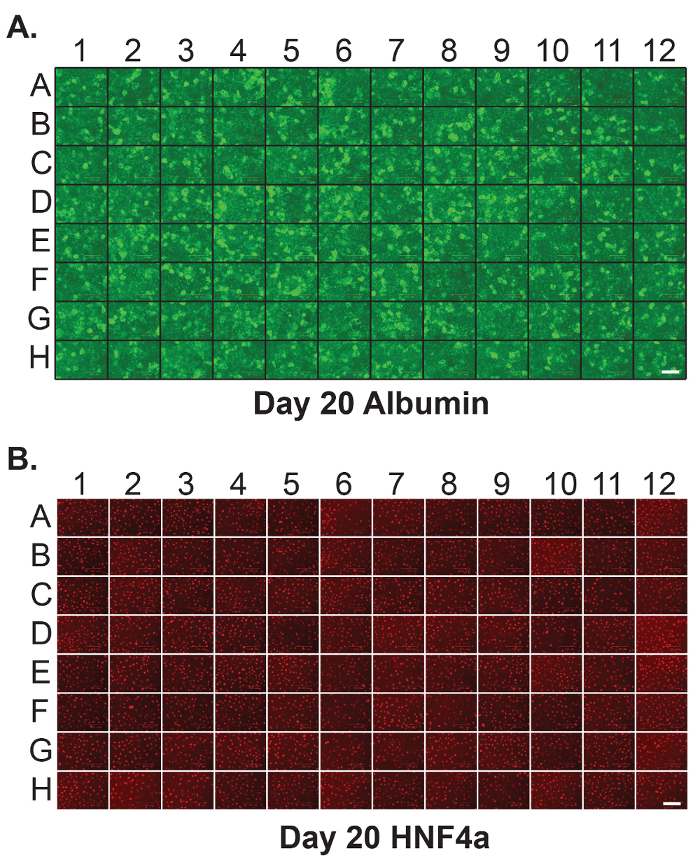
Figure 2: Homogenous hepatic differentiation of human iPSCs in 96-well plates. Immunostaining was performed on each individual well of a 96-well plate containing iPSC-derived hepatocytes at day 20 of differentiation to detect (A) Albumin and (B) HNF4a. Scale bar = 100 µm. Please click here to view a larger version of this figure.
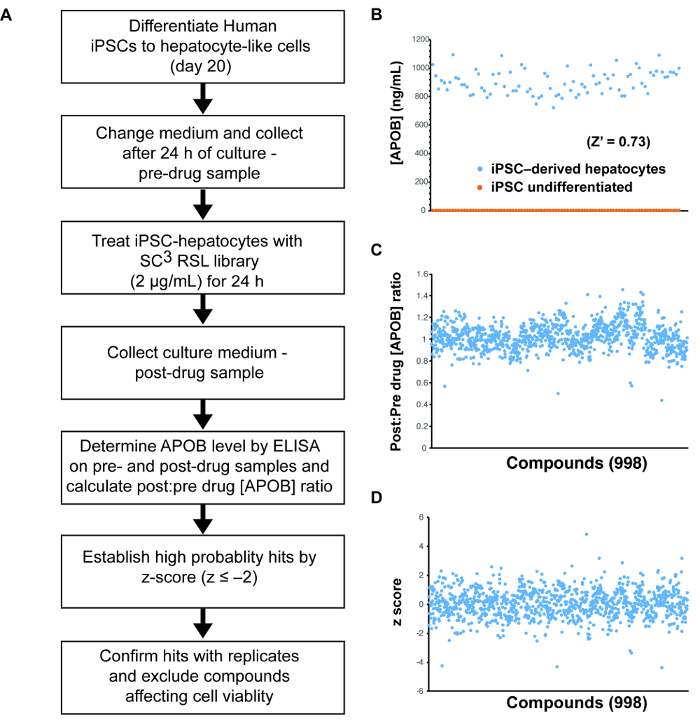
Figure 3: Small molecule screening using human iPSC-derived hepatocytes. (A) Schematic overview of the approach used to identify compounds that can reduce the levels of ABOB in the medium of iPSC-derived hepatocytes. (B) Graph showing theresults of an ELISA assay to detect the level of APOB in the medium of 87 wells (n = 87) of iPSC-derived hepatocytes (blue) and/or undifferentiated iPSCs (orange). These data were used to calculate a Z-factor (Z' = 0.73). (C) Graph showing the post-drug: pre-drug ratios of the concentration of APOB in the culture medium of iPSC-derived hepatocytes treated for 24 h with 998 compounds from the SC3 RSL set. (D) Graph showing z-scores of the data presented in (C). Please click here to view a larger version of this figure.
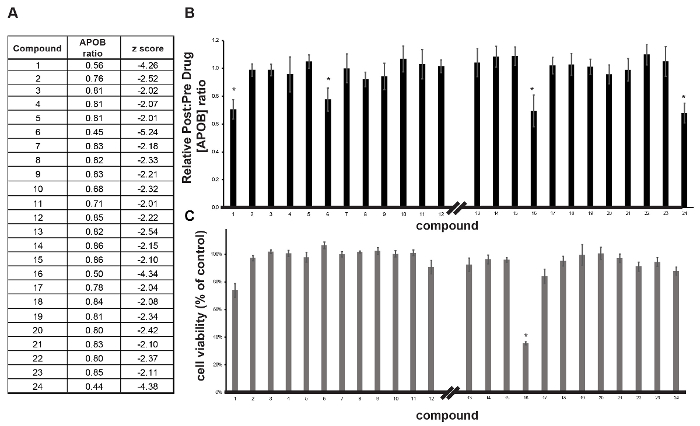
Figure 4: Validation of primary hits. (A) Table showing compounds identified in the primary screen as being able to reduce APOB. (B, C) Bar graphs showing the impact of compounds on post-drug: pre-drug APOB concentration ratio (B) and cell viability (C) in iPSC-derived hepatocytes treated with compounds in follow-up analysis. Error bars reflect ± SEM (n = 4 biological replicates); * p ≤0.05. Please click here to view a larger version of this figure.
