Evaluation of the method for identification of protease specificity by model protease
The efficiency of this system was evaluated using TPCK-trypsin and V8 protease, whose substrate specificities were obtained. TPCK-trypsin and V8 protease were estimated to cleave the amino acid sequence at the C-terminal of the lysine and arginine residues, and at the carboxy side of the aspartic acid and glutamic acid residues, respectively. Table 1 shows the theoretical molecular weight of the substrate and digested fragments cleaved by specific proteases. Substrate digestion using TPCK-trypsin produced cleaved fragments, two of which could be detected at 839.81 Da and 796.76 Da (Figure 2A). The molecular weights of these fragments were closely in line with the theoretical molecular weights of the fragments cleaved from the C-terminal of lysine and arginine, respectively. In addition, the V8 protease converted precursor substrates to their cleaved form, with molecular weights of 769.78 Da and 755.62 Da (Figure 2B), which matched the theoretical m/z of glutamic acid and aspartic acid, respectively. These results indicate that this method could be used to successfully identify protease specificity with MALDI-TOF mass spectrometry.
Evaluation of substrate specificity at different pH levels using mouse lung extracts
One of the advantages of this technique was that a large number of samples can be simultaneously processed. This study demonstrated that substrate specificity changed according to pH level (Figure 3). In acidic conditions (Figure 3B; pH 5), the molecular weight of a cleaved fragment was 770.13 Da and 826.66 Da. These results indicated that the lung tissue extract contained proteases with the ability to cleave at the C-terminus of glutamic acid and tryptophan. As the pH level gradually increased, the peaks of fragments cleaved by glutamic acid and tryptophan gradually decreased, whereas peaks of fragments cleaved by arginine and lysine gradually increased (Figure 3C, D; pH 7 and 9). These results indicated that the lung extract contained two or more proteases that had different optimum pH and cleavage specificities.
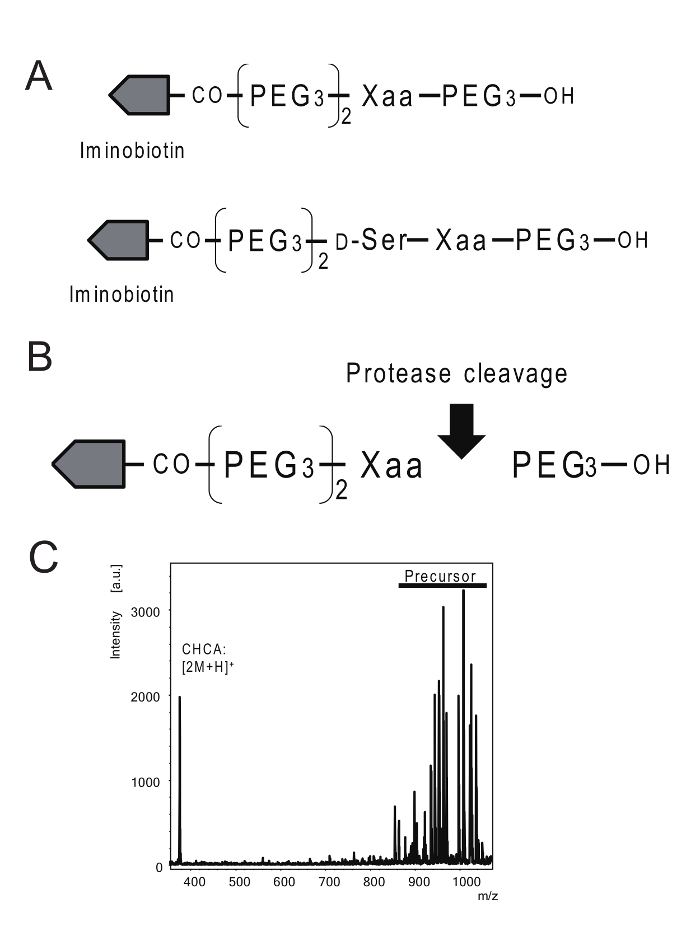
Figure 1: Schema for detection of protease specificity. (A) The structure of iminobiotin-labeled substrate for protease specificity. (B) The substrates had iminobiotin, a polyethylene glycol spacer, and a cleavable amino acid site. (C) The spectra of iminobiotin-labeled substrates. Please click here to view a larger version of this figure.
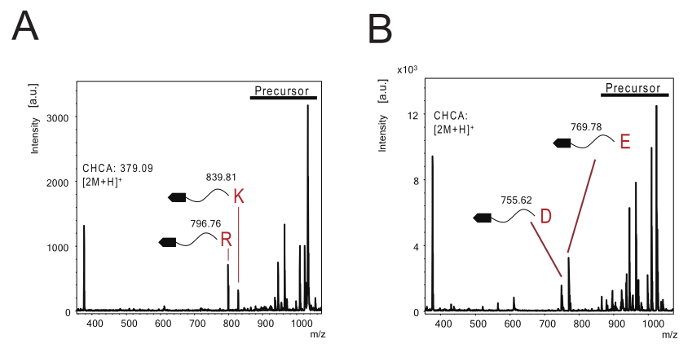
Figure 2: Evaluation of the method to determine protease specificity using a purified enzyme. (A) The spectra for iminobiotin-labeled substrates treated with TPCK-trypsin. The 839.81 and 796.76 peaks matched the theoretical molecular weight of fragments generated by cleavage at the C-terminal side of lysine and arginine, respectively. (B) The spectra for iminobiotin-labeled substrates treated with V8 protease. The 769.78 and 755.62 peaks matched the theoretical molecular weight of fragments generated by cleavage at the C-terminal side of glutamic acid and aspartic acid, respectively. Please click here to view a larger version of this figure.
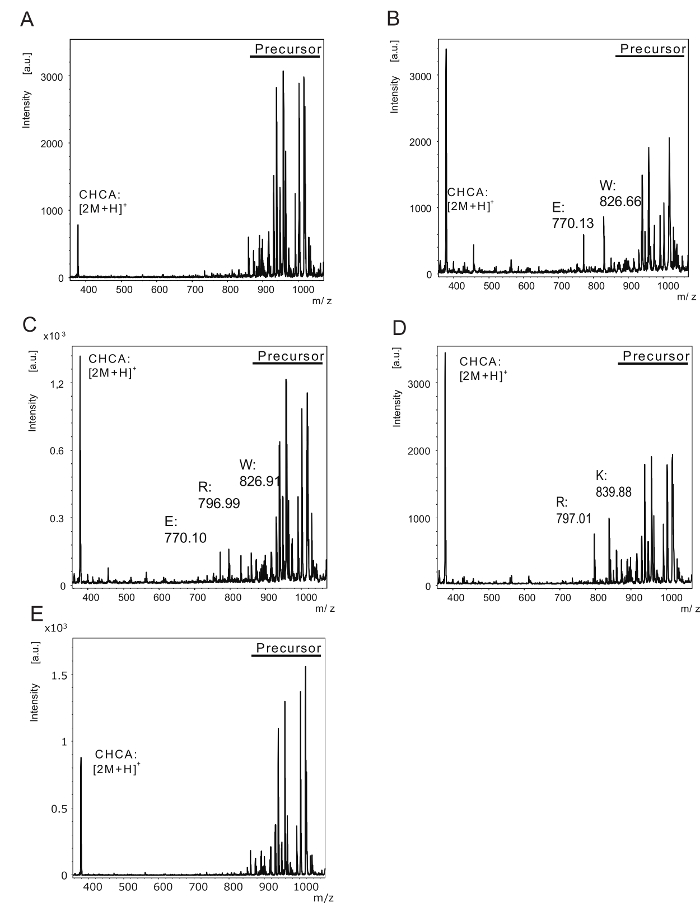
Figure 3: Evaluation of the method to determine protease specificity using tissue extracts. The spectra for iminobiotin-labeled substrates treated with a lung tissue extract. These spectra show peaks obtained at (A) pH 3, (B) pH 5, (C) pH 7, and (D) pH 9. (E) The iminobiotin-labeled substrates incubated with the tissue extract inactivated with heat treatment to evaluate non-specific digestion. Please click here to view a larger version of this figure.
| X | Precursor | Cleaved form |
| G | 886.58 | 697.58 |
| A | 900.60 | 711.60 |
| S | 916.59 | 727.59 |
| P | 926.61 | 737.61 |
| V | 928.63 | 739.63 |
| T | 930.61 | 741.61 |
| C* | 1003.57 | 814.57 |
| I* | 1013.64 | 824.64 |
| L | 942.64 | 753.64 |
| N* | 1014.60 | 825.60 |
| D | 944.59 | 755.59 |
| Q | 957.62 | 768.62 |
| K* | 1028.65 | 839.65 |
| E | 958.60 | 769.60 |
| M* | 1031.60 | 842.60 |
| H | 966.62 | 777.62 |
| F | 976.63 | 787.63 |
| R | 985.66 | 796.66 |
| Y | 992.62 | 803.62 |
| W* | 1015.64 | 826.64 |
| *: D-Ser-Xaa form | ||
Table 1: The theoretical molecular weight of iminobiotin-labeled substrates and the expected molecular weight of protease fragments. Asterisks (*) indicate addition of D-serine between the polyethylene spacer and the cleavable amino acid.
| Xaa: Gly, Ala, Ser, Pro, Val, Thr, Leu, Asp, Gln, Glu, His, Phe, Arg, Tyr | |||
| Stage | Fmoc-compound | ||
| 1 | Fmoc-mini-PEG3-OH | ||
| 2 | Fmoc-Xaa-OH | ||
| 3 | Fmoc-mini-PEG3-OH | ||
| 4 | Fmoc-mini-PEG3-OH | ||
| 5 | Iminobiotin | ||
| Xaa: Cys, Ile, Asn, Lys, Met, Trp | |||
| Stage | Fmoc-compound | ||
| 1 | Fmoc-mini-PEG3-OH | ||
| 2 | Fmoc-Xaa-OH | ||
| 3 | Fmoc-D-Ser(tBu)-OH | ||
| 4 | Fmoc-mini-PEG3-OH | ||
| 5 | Fmoc-mini-PEG3-OH | ||
| 6 | Iminobiotin | ||
Table 2: Sequential list of reagents used for substrate synthesis.
| pH | composition | ||
| 9 | 50 mM Tricine containing 0.1 M NaCl and 10 mM CaCl2 | ||
| 7 | 50 mM Tris-HCl containing 0.1 M NaCl and 10 mM CaCl2 | ||
| 5 | 50 mM sodium acetate containing 0.1 M NaCl and 10 mM CaCl2 | ||
| 3 | 50 mM citric acid containing 0.1 M NaCl and 10 mM CaCl2 | ||
Table 3: Compositions of the buffer solutions used.
