Production of Elastin-like Protein Hydrogels for Encapsulation and Immunostaining of Cells in 3D
Summary
Recombinant protein-engineered hydrogels are advantageous for 3D cell culture as they allow for complete tunability of the polymer backbone and therefore, the cell microenvironment. Here, we describe the process of recombinant elastin-like protein purification and its application in 3D hydrogel cell encapsulation.
Abstract
Two-dimensional (2D) tissue culture techniques have been essential for our understanding of fundamental cell biology. However, traditional 2D tissue culture systems lack a three-dimensional (3D) matrix, resulting in a significant disconnect between results collected in vitro and in vivo. To address this limitation, researchers have engineered 3D hydrogel tissue culture platforms that can mimic the biochemical and biophysical properties of the in vivo cell microenvironment. This research has motivated the need to develop material platforms that support 3D cell encapsulation and downstream biochemical assays. Recombinant protein engineering offers a unique toolset for 3D hydrogel material design and development by allowing for the specific control of protein sequence and therefore, by extension, the potential mechanical and biochemical properties of the resultant matrix. Here, we present a protocol for the expression of recombinantly-derived elastin-like protein (ELP), which can be used to form hydrogels with independently tunable mechanical properties and cell-adhesive ligand concentration. We further present a methodology for cell encapsulation within ELP hydrogels and subsequent immunofluorescent staining of embedded cells for downstream analysis and quantification.
Introduction
Over the past century, two-dimensional (2D) tissue culture has developed into an integral toolset for studying fundamental cell biology in vitro. In addition, the relatively low-cost and simple protocols for 2D cell culture have led to its adoption across many biological and medical disciplines. However, past research has shown that traditional 2D platforms can lead to results that deviate markedly from those collected in vivo, causing precious time and funding wasted for clinically oriented research1,2,3. We and others hypothesize that this discrepancy may be attributed to the lack of native biochemical and biophysical cues provided to the cells cultured on 2D surfaces, which can be necessary for optimal proliferation and maturation of various cell types.
To address these limitations and help bridge the gap between 2D in vitro and in vivo studies, researchers have developed three-dimensional (3D) hydrogel platforms for cell-encapsulation1,4,5,6. Hydrogels are ideal materials to recapitulate the endogenous microenvironment of the extracellular matrix (ECM) in vivo due to their tissue-like mechanical properties and water-swollen structure that enables rapid transport of nutrients and signaling factors7,8. Furthermore, 3D hydrogels can be designed to have independent control over the mechanical and biochemical properties of the scaffold. Both matrix mechanics9,10,11,12 and cell-adhesive ligands13,14,15 are well-known to influence cell behavior in vitro and in vivo. Thus, 3D hydrogels with tunable properties offer a platform to study the causal relationships between cells and their microenvironment. Criteria for an ideal 3D hydrogel matrix include simple, non-cytotoxic cell-encapsulation as well as independent tunability of physiologically relevant mechanical properties and mimics of native cell-adhesive motifs.
Both synthetic (e.g., polyethylene glycol, polylactic acid, poly(glycolic acid)) and naturally-derived (e.g., alginate, collagen, Matrigel) hydrogels have advantages over 2D in vitro culture platforms; however, they also have significant shortcomings which limit their applicability. First, many synthetic and naturally-derived platforms require harsh crosslinking conditions that can be potentially toxic to mammalian cells, leading to decreased cell viability7. Additionally, many synthetic platforms lack native bioactivity and need to be functionalized through secondary chemical reactions, which can add increased cost and complexity16. Finally, while naturally-derived materials typically contain intrinsic bio-active domains, they are often plagued by high batch-to-batch variability and often are limited to forming relatively weak gels7,17.
Recombinant protein engineering presents a unique toolset for materials design by allowing explicit control over protein sequence and, by extension, the potential mechanical and biochemical properties of the final hydrogel scaffold18. Additionally, by leveraging the well-known biological machinery of Escherichia coli (E. coli) to express proteins, materials can be produced cost-effectively and consistently with limited inter- and intra-batch variability. The elastin-like protein (ELP) presented here has three engineered domains: (1) a T7 and His6 tag that allows for labeling via fluorescently tagged antibodies, (2) an 'elastin-like' region that confers elastic mechanical properties and allows for chemical crosslinking, and (3) a 'bio-active' region that encodes for cell-adhesive motifs.
Our elastin-like region is based on the canonical (Val-Pro-Gly-Xaa-Gly)5 elastin sequence where four of the 'Xaa' amino acid sites are isoleucine (Ile), but could be designed to be any amino acid except proline. This sequence endows recombinant ELPs with lower critical solution temperature (LCST) behavior that can be exploited for simple purification post-expression via thermal cycling19,20. This LCST property can be tuned to thermally aggregate at different temperatures by modifying the guest 'Xaa' residue21,22.
Here, the 'Xaa' position on one of the five elastin-like repeats has been replaced with the amine-presenting lysine (Lys) amino acid, which is utilized for hydrogel crosslinking. Our previous work has shown non-cytotoxic and robust crosslinking via reaction with the amine-reactive crosslinker tetrakis(hydroxymethyl)phosphonium chloride (THPC)23. By varying overall protein content and crosslinker concentration, we are able to produce hydrogels that can be tuned to span a physiologically relevant stiffness range (~0.5-50 kPa)9,23,24. In addition to tuning mechanical properties, cell adhesion within the hydrogel results from the integration of canonical cell-adhesive domains within the backbone of the ELP protein. For example, the incorporation of the extended fibronectin-derived 'RGDS' amino acid sequence allows for cell adhesion and conformational flexibility, while the scrambled, non-binding 'RDGS' variant restricts cell-matrix adhesion24. By modulating the ratio of cell-adhesive to non-adhesive proteins as well as the total protein concentration, we are able to effectively produce hydrogels which span a wide range of ligand concentration. Resultantly, we have developed a hydrogel platform with decoupled biochemical and biophysical properties, which can be independently tuned for optimal 3D culture of various cell types.
In addition to matrix stiffness and adhesive ligand tunability, recombinant hydrogels offer the capability to design specific material degradation profiles, which is necessary for cell spreading, proliferation, and migration within a 3D context4,9. This degradation is afforded by cell secretion of proteases that specifically target either the extended 'RGDS'9 or elastin-like sequence25. ELP hydrogels have also been shown to support the subsequent biochemical assays that are necessary for studying cell viability and function including immunocytochemistry as well as DNA/RNA/protein extraction for quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and Western blot9. ELP variants have also been used in a number of in vivo models and are known to be well tolerated by the immune system26.
Taken together, ELP as a material platform for cell-encapsulation studies boasts a wide variety of benefits compared to synthetic or naturally-derived material platforms, which often lack the same degree of biochemical and biophysical tunability and reproducibility. Additionally, ELP's simple and non-cytotoxic use with a wide variety of cell types (e.g., chick dorsal root ganglia14,24, murine neural progenitor cells9, human mesenchymal stem cells27, bovine neonatal chondrocytes28, human endothelial cells29,30) allows for a more physiologically relevant model of the endogenous 3D ECM compared to 2D cell culture. Herein, we present a protocol for the expression of recombinantly-derived, ELPs for the use as a tunable hydrogel platform for 3D cell encapsulation. We further present the methodology for down-stream fluorescent labeling and confocal microscopy of encapsulated cells.
Protocol
1. ELP Expression Protocol
- Day 1: Growing the starter colony
- Prepare ampicillin and chloramphenicol agar plates by autoclaving 25 g of Luria broth and 15 g of agar per 1 L of ultrapure water. Once the solution has cooled to ~60 °C, add 1 mL of ampicillin stock (100 mg/mL in ultrapure water) and 1 mL of chloramphenicol stock (34 mg/mL in 70% ethanol) to 1 L of agar solution for final concentrations of 100 µg/mL and 34 µg/mL, respectively. Transfer 20 mL of final solution to 10 cm Petri dishes with a serological pipette and allow the agar to solidify. Wrap the Petri dishes with parafilm and store at 4 °C.
NOTE: Petri dishes can be stored at 4 °C for up to two weeks. - Streak a small sample of BL21(DE3)pLysS E. coli from a pre-made bacterial stock containing a pET15b vector encoding the ELP of interest on an ampicillin and chloramphenicol agar plate.
NOTE: The ampicillin and chloramphenicol select for bacteria containing both the pET15b and pLysS vectors, respectively. - Place the streaked plate upside down in an incubator at 37 °C. Allow the bacterial colonies to grow overnight.
NOTE: Do not incubate the plates for longer than 16 h as ampicillin will degrade and the colonies that do not carry ampicillin resistance can form.
- Prepare ampicillin and chloramphenicol agar plates by autoclaving 25 g of Luria broth and 15 g of agar per 1 L of ultrapure water. Once the solution has cooled to ~60 °C, add 1 mL of ampicillin stock (100 mg/mL in ultrapure water) and 1 mL of chloramphenicol stock (34 mg/mL in 70% ethanol) to 1 L of agar solution for final concentrations of 100 µg/mL and 34 µg/mL, respectively. Transfer 20 mL of final solution to 10 cm Petri dishes with a serological pipette and allow the agar to solidify. Wrap the Petri dishes with parafilm and store at 4 °C.
- Day 2: Preparation of the starter culture and expression media
- Remove the E. coli culture from the incubator. Parafilm the plate and store for a maximum of 4 days at 4 °C.
- Prepare a starter culture flask (250 mL) and expression culture flasks (12x 1 L) and autoclave. For 1 L of expression media, add 47.6 g of terrific broth and 4 mL of glycerol to 1 L of ultrapure water in a 2 L baffled culture flask, and cap with aluminum foil.
NOTE: Typical yields are 60-100 mg/L protein of expression media. - Load the autoclaved starter into pre-warmed, 37 °C shaking incubator and incubate without agitation.
- Add 250 µL of sterile-filtered (0.22 µm filter) ampicillin stock (100 mg/mL in ultrapure water) to the starter culture for a final concentration of 100 µg/mL. Immediately begin agitating the starter culture at 250 rpm.
NOTE: Chloramphenicol is only used for colony selection on agar plates and is not included for liquid cultures. - Inoculate the starter culture by adding a single ELP plasmid-containing E. coli colony from the streaked plate and allow the starter culture to shake at 37 °C for 16 h.
- Place the expression culture media flasks into pre-warmed, 37 °C shaking incubators and incubate overnight without agitation so that the flasks are ready to inoculate the next morning.
- Day 3: Inducing protein expression in E. coli
- Make fresh, sterile-filtered ampicillin stock (100 mg/mL in ultrapure water). Add 1 mL of ampicillin stock to each flask of expression media for a final concentration of 100 µg/mL.
- Begin agitating the expression media at 250 rpm.
- Take a 2 mL sample of the media from any expression flask and add to a cuvette as a blank for an optical density reading at 600 nm (OD600).
- After the completion of 16 h starter culture incubation, inoculate each expression flask by transferring 20 mL of the starter culture to each expression flask via a serological pipette.
- After the completion of 1 h agitation, measure the OD600 of one of the expression flasks. Following this step, measure the OD600 every 20 min, checking a different flask each time.
- At an OD600 of 0.6, reduce the temperature of the shakers containing the expression flasks to 32 °C.
- Check the OD600 every 10 min. At an OD600 of 0.8, induce the expression by adding 1 mL of 1 M, sterile-filtered β-isopropyl thiogalactoside (IPTG) in ultrapure water to each expression flask.
- Allow the E. coli in expression flasks to express for 7 h.
- 20 min prior to the end of expression, pre-cool a large floor centrifuge to 4 °C.
- Collect all 12 L of expression media into individual centrifuge containers and balance.
- Centrifuge the expression media at >12,000 x g for 15 min at 4 °C using a floor centrifuge.
- Pour off the supernatant from each centrifuge container. Using a spatula, collect the cell pellets into a pre-weighed zip lock bag.
- Re-suspend the pellet in sterile-filtered TEN buffer (100 mL of buffer per 25 g of pellet) and remove any excess air bubbles by massaging the pellet. For 1 L of TEN buffer, add 5.8 g of sodium chloride, 1.21 g of tris base, and 0.37 g of ethylenediamine tetraacetic acid (EDTA) disodium salt dihydrate to 900 mL of ultrapure water. Adjust the pH to 8.0 and bring to 1 L. For this step, pH strips are sufficient.
- Place the zip-lock bag containing the re-suspended cell pellet into a secondary container and freeze at -80 °C overnight.
- Day 4-6: Rupturing the bacterial cell wall via freeze-thaw cycles
- Remove the frozen pellet from the freezer and allow it to slowly thaw at 4 °C with gentle agitation using an orbital shaker.
- Allow the pellet to thaw until some liquid is present. Add ~30-40 mg of deoxyribonuclease I (DNase) to thawed lysate. Additionally, add 1 mL of 100 mM phenylmethylsulphonyl fluoride (PMSF; a protease inhibitor) in isopropanol per 100 mL of cell lysate. Allow the lysate to shake overnight.
NOTE: Adding the DNase and PMSF is only necessary for the first freeze-thaw cycle.
CAUTION: PMSF is toxic if inhaled. A face mask should be used when handling PMSF powder. - Once the cell lysate is completely thawed, freeze the lysate at -80 °C overnight, or until completely frozen.
- Repeat the freeze-thaw procedure for a total of three cycles. Leave the lysate thawed at 4 °C after the last freeze-thaw. Store 100 µL of raw lysate for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis at the conclusion of purification.
- After the last thaw, adjust the pH of the thawed lysate to 9.0 using 1 M NaOH. For this step, pH strips are sufficient. Incubate at 4 °C for at least 1 h (overnight is appropriate) on an orbital shaker.
- Day 7-9: ELP purification via cold and hot spin thermal cycling
- Cool the floor centrifuge to 4 °C 20 min prior to centrifugation.
- Aliquot and balance the thawed lysate into centrifuge containers.
- Centrifuge the samples at >15,000 x g for 1 h at 4 °C. Store 100 µL of the supernatant for SDS-PAGE analysis after the completion of purification.
NOTE: After centrifuging, the ELP protein should remain in the supernatant due to its high solubility in water at temperatures below its LCST (<32 °C). - Transfer the supernatant to new centrifuge containers and balance appropriately.
- Add sodium chloride (NaCl) in three parts to a final concentration of 1 M (5.84 g of NaCl for every 100 mL of the supernatant). Ensure that NaCl is added in three parts to allow for sufficient dissolution.
- Agitate for 3 h at 37 °C at 250 rpm in a shaking incubator.
- 1 h before the end of agitation, pre-warm a floor centrifuge to 37°C.
- Centrifuge at >15,000 x g for 1 h at 37 °C. Store 100 µL of the supernatant for SDS-PAGE analysis after the completion of purification and discard the remaining supernatant.
NOTE: After centrifuging, the ELP protein should be pelleted due to its low solubility in water at temperatures above its LCST (>32 °C). - Re-suspend the pellet by adding 10 mL of autoclaved, ultrapure water per 1 g of pellet. Use a metal spatula to mash the pellet to aid in the dissolution of the protein.
- Adjust the pH of the thawed lysate to 9.0 using 1 M NaOH. For this step, pH strips are sufficient.
- Agitate overnight at 4 °C on an orbital shaker.
- Repeat the cold and hot spin thermal cycling procedure for a total of three cycles (i.e., repeat steps 1.5.1 through 1.5.11 for three total cycles).
- Day 10-15: ELP dialysis and lyophilization
- Centrifuge the re-suspended pellet in a pre-chilled centrifuge at >15,000 x g for 1 h at 4 °C. Store 100 µL of the supernatant for SDS-PAGE analysis after the completion of purification.
- Desalt the protein solution by dialyzing the remaining supernatant in a 3.5 kDa dialysis membrane against 4 L of pre-chilled ultrapure water at 4 °C. Change the dialysis water twice a day for a total of 6 times over 3 days.
NOTE: Typical supernatant volumes are between 5 and 30 mL. - Centrifuge the dialyzed solution in a pre-cooled centrifuge at >15,000 x g for 1 h at 4 °C. Store 100 µL of the supernatant for SDS-PAGE analysis at the end of the purification.
- Freeze the resulting supernatant at -80 °C in pre-weighed conical tubes.
- Lyophilize the frozen solution for 3 days and mass the final product to determine the protein yield.
- Parafilm the tubes containing the final lyophilized ELP product and store at 4 °C.
- Run SDS-PAGE to determine the purity of the protein.
NOTE: Protocols for SDS-PAGE will vary based on specific agarose gel conditions. For our experiments, final lyophilized protein was dissolved to a concentration of 0.5 mg/mL in deionized water and run at 140 V for 70-100 min in a 12% (w/v) acrylamide gel under denaturing conditions.
2. Cell Encapsulation in 3D Elastin-like Protein Hydrogels
- Preparation of silicone molds
- Use a biopsy punch with the desired diameter to create holes in a 0.5 mm thick silicone sheet and cut out a square around each hole. Repeat for the number of molds desired.
NOTE: The diameter and thickness of the molds can be adjusted for the particular application and cell culture conditions. In practice, for cell cultures of 50 106 cells/mL, 0.5 mm thick molds with 4 mm and 5 mm diameters are recommended for sufficient DNA/RNA and protein extraction, respectively. For immunostaining, a 2 mm biopsy punch can be used to instead create three adjacent holes per square mold so that three replicates can be stained per well. - Remove the plastic wrap on each side of the individual mold with tweezers.
NOTE: Avoid contact with the exposed silicone surface as contamination can decrease future plasma bonding efficiency. - Using tweezers, arrange the same number of bare silicone molds and glass coverslips (No. 1, 12 mm diameter) in alternating rows on the inverted lid of a 48-well plate. Once completed, cover the lid of the 48-well plate to avoid contamination.
- Oxygen plasma treat the entire 48-well plate lid (i.e., molds and glass slides). Immediately after, use forceps to invert the silicone mold onto the adjacent glass coverslip. Press firmly on the mold to ensure bonding. Let the molds incubate at room temperature for 1 h.
NOTE: The duration of oxygen plasma treatment will vary depending on the instrument used. Typical conditions for our instrument are an operating gas pressure window between 0.3-4 mbar, oxygen gas flow at 20 cm3/min, and sample exposure to plasma for 10-20 s. - Sterilize the molds by autoclaving. Store the molds at room temperature in a sterile environment until use.
NOTE: The molds can be stored at this stage indefinitely.
- Use a biopsy punch with the desired diameter to create holes in a 0.5 mm thick silicone sheet and cut out a square around each hole. Repeat for the number of molds desired.
- Preparation of elastin-like protein stock solution
- Remove lyophilized ELP from 4 °C storage. Warm the protein to room temperature before opening the tube to ensure no condensation builds on the protein over repeated use.
- Dissolve ELP in Dulbecco's phosphate-buffered saline (DPBS) at 4 °C with constant agitation (i.e., spinning) overnight.
NOTE: ELP stock solution concentration will be further diluted with the addition of crosslinker solution at a user-defined volumetric ratio. For example, for a 3% (w/v) final ELP concentration, prepare a 3.75% (w/v) ELP stock solution that will be diluted at a 4:1 volumetric ratio of ELP solution:crosslinker solution. Adjust the concentration as necessary for desired application. - Sterile filter ELP stock solution using a 0.22 µm syringe filter. Store ELP on ice when not in use.
- In the biosafety cabinet, transfer the sterile molds using sterile tweezers to a 24-well tissue-culture plate.
- Preparation of THPC stock solution
- Dilute THPC in DPBS just prior to use. Adjust the concentration of THPC solution according to the final ELP concentration and desired crosslinking ratio.
NOTE: For the protocol, a final ELP concentration of 3% (w/v) will be made by mixing the ELP stock solution with the THPC solution at a volumetric ratio of 4:1. Adjust as necessary. - Add 2.6 µL of THPC solution (80% in water) to 997.4 µL of DPBS.
NOTE: This concentration of THPC corresponds to a 1:1 stoichiometric ratio of hydroxy methyl groups on THPC and primary amines on the ELP protein when mixing solutions at the described 4:1 volumetric ratio for a final 3% (w/v) ELP hydrogel. The THPC solution is viscous and droplets of solution may stick to the side of the pipette tip. For accurate concentrations, avoid such droplets when diluting into DPBS. Purge the THPC stock container with nitrogen gas to prevent oxidation of the phosphine and inactivation of the crosslinker. - Vortex the solution to mix and keep on ice.
- Sterile filter the THPC stock solution using a 0.22 µm syringe filter. Dilute the THPC stock solution further with sterile DPBS to achieve lower stoichiometric crosslinking ratios (e.g., 0.5:1 or 0.75:1).
NOTE: THPC is oxygen-sensitive, and the diluted solution should be used within a few hours after preparation.
- Dilute THPC in DPBS just prior to use. Adjust the concentration of THPC solution according to the final ELP concentration and desired crosslinking ratio.
- Dissociate the cells by incubation with Trypsin-EDTA into a single-cell suspension, pellet the cells, and count the cells re-suspended in medium using a hemocytometer.
NOTE: Exact protocols for this step will depend heavily on desired cell type and application. For the neural progenitor cells used throughout this protocol, a 0.025% Trypsin-EDTA incubation at room temperature for 1.5 min was conducted. The cells were pelleted at 200 x g for 2 min. For increased cell viability, cells should be suspended in normal medium conditions. Typical cell densities used for cell encapsulation range from 1 – 50 106 cells/mL of final hydrogel volume. Adjust as necessary. - Aliquot the desired number of cells into a sterile 1.5 mL centrifuge tube.
- Centrifuge the cells at ~200 x g for 3 min. Carefully, aspirate the supernatant and keep the cell pellet on ice.
NOTE: It is critical to completely aspirate all the supernatant to mitigate the further dilution of the ELP and THPC crosslinker. Specific centrifugation speeds will vary with cell type. - Re-suspend the cell pellet in the ELP stock solution such that the volume is 80% of the final volume (assuming a 4:1 ratio of ELP solution:THPC solution). Pipette mix 20-25 times to produce a homogenous mixture of the cells and ELP.
NOTE: Avoid gripping the bottom of the tube to mitigate temperature increase and subsequent phase transition of ELP. Each 2 mm mold with three replicates requires 7.5 µL of final volume (i.e., 6 µL of ELP stock solution and 1.5 µL of THPC stock solution) divided equally into the 3 holes (i.e., 2.5 µL final volume per hole). For the 4 and 5 mm molds, a final volume of 7.5 µL and 15.5 µL is required, respectively. - Add THPC stock solution to the cell/ELP suspension for the remaining 20% final volume. Pipette mix 20-25 times to produce a homogenous mixture.
- Immediately pipette the corresponding final volumes of cell/ELP/THPC mixture into each mold by a circular motion. Repeat for all molds.
- Incubate the samples at room temperature for 15 min, followed by an additional incubation at 37 °C for 15 min.
NOTE: The first incubation period will help facilitate initial crosslinking of the hydrogel before increasing the temperature to that above the ELP's LCST and inducing its thermal phase segregation. - Slowly add 750 µL of warm cell culture medium to each well of the 24-well plate, avoiding disrupting the gels.
- Incubate the hydrogels at 37 °C for 7 days.
NOTE: Full medium changes are recommended every 1-2 days depending on cell type. To limit the stress on the gel, use a glass Pasteur pipette affixed with a 200 µL pipette tip when aspirating medium from the well.
3. Immunocytochemistry of Cells in 3D ELP Hydrogels
- Prepare the fixation solution by mixing 10 mL 16% (w/v) paraformaldehyde (PFA) in 30 of mL DPBS. Warm the solution to 37 °C.
- Aspirate the medium from the 24-well plate and gently wash with 1 mL of DPBS.
- Add 750 µL of the fixation solution to each well and incubate at 37 °C for 30 min. Do not use the tissue culture incubator to avoid contamination of other cultures with PFA vapor.
- Carefully aspirate the fixation solution from each well, discarding the PFA to an appropriate hazardous waste container.
CAUTION: Exposure to PFA can cause skin and eye irritation. Wear gloves/safety glasses and work in a chemical fume hood. - Add 1 mL of DPBS to each sample. Immediately aspirate the DPBS and discard into PFA waste container.
- Wash the samples twice with 1 mL of DPBS for 10 min each.
NOTE: Samples can be stored in DPBS at 4 °C for up to a week after sealing the plate with Parafilm. - Permeabilize each sample with 750 µL of permeabilization solution (100 mL of DPBS and 0.25 mL of Triton X-100; PBST) for 1 h at room temperature on a rocker at 15 rpm.
- Aspirate the PBST and add 750 µL of blocking solution (95 mL of PBS, 5 g of bovine serum albumin (BSA), 5 mL of serum, and 0.5 mL of Triton X-100) to each sample. Incubate at room temperature for 3 h on a rocker at 15 rpm.
NOTE: The blocking solution should contain serum from the host species in which the secondary antibodies were raised (e.g., goat, donkey) and sterile-filtered through a 0.22 µm filter prior to use. - Prepare the antibody dilution solution (97 mL of DPBS, 2.5 g of BSA, 2.5 mL of serum (from same host species as in step 3.8), and 0.5 mL of Triton X-100). Dilute the primary antibody using the antibody dilution solution and add 500 µL of the solution to each sample. Seal the plate with Parafilm and incubate overnight at 4 °C on a rocker.
NOTE: Nestin and Sox2 primary antibodies were diluted 1:400 in antibody dilution solution from the manufacturers' original concentration. - Aspirate the antibody solution from each sample and wash the samples with PBST for 60 min at room temperature on a rocker at 15 rpm. Repeat the wash step 3 times.
- Dilute the secondary antibodies and 5 mg/mL stock DAPI (1:2,000) using the antibody dilution solution and add 500 µL of the solution to each sample. Cover the 24-well plate with aluminum foil and incubate overnight at 4 °C on a rocker.
NOTE: As the secondary antibodies are light sensitive, the samples must be protected from photobleaching for all subsequent steps. Goat anti-mouse and goat anti-rabbit secondary antibodies were diluted 1:500 in antibody dilution solution from the manufacturers' original concentration. - Aspirate the antibody solution from each sample and wash the samples with PBST for 30 min at room temperature on the rocker. Repeat the wash step 3 times.
- Position a drop of hard-set mounting medium onto the surface of a glass slide. Using forceps, remove excess solution from the mold by lightly blotting the edge of the mold on a paper towel. Carefully place the mold upside down onto the mounting medium and avoid introducing bubbles.
- Allow the mounting medium to harden for 48 h at room temperature. Store the samples at room temperature or 4 °C. Allow the mounting medium to fully set for 48 h before imaging as refractive index of the medium will change over the course of hardening. Seal the samples to the glass cover slide with clear nail polish to mitigate contamination or sample movement.
- Image the samples using a confocal microscope.
Representative Results
The ELPs used in this protocol are comprised of five regions: a T7 tag, His6 tag, enterokinase (EK) cleavage site, a bio-active region, and an elastin-like region (Figure 1). The T7 and His6 tags allow for easy identification through standard Western blot techniques. Introduction of the EK cleavage site allows for the enzymatic removal of the tag region, if needed. The bio-active region encodes for the extended, fibronectin-derived cell-adhesive ('RGDS') or non-adhesive ('RDGS') sequences. Lastly, the central repeat of the elastin-like region contains a lysine group at the guest residue site which enables crosslinking via THPC while the flanking repeats contain isoleucine to achieve an LCST of ~32 °C31.
Post expression, SDS-PAGE or Western blot can be used to visualize the molecular weight and confirm the identity of ELPs that contain antibody tags, such as T7 (MASMTGGQQMG) or His6 (HHHHHH) (Figure 2). Successful expression under controlled conditions yields a highly homogeneous product represented by the presence of a single dark band at the approximate molecular weight (~37 kDa) of these proteins using both SDS-PAGE (Figure 2A) and Western blot (Figure 2B).
In uncontrolled conditions, the presence of lower molecular weight bands on a Western blot suggests that some fraction of the proteins was not completely translated and/or were degraded after expression (Figure 2C). Specifically, the masses here are evenly spaced by ~9 kDa which roughly corresponds to the weight of one bio-active region and three elastin-like regions (a 'repeat'), or approximately a fourth of the target protein. These smaller protein fragments are usually present when the expression is carried out at a higher temperature (>32 °C) as in Figure 2C. The presence of these lower molecular weight proteins can lead to unpredictable mechanical properties. Thus, regular screening post expression is recommended to ensure a high quality final product.
The mechanical stiffness of ELP-based hydrogels can be modified by manipulating the concentration of ELP or the ratio of THPC reactive groups:ELP primary amines. Concurrently, the concentration of cell-adhesive ligands can be tuned by changing the ratio of ELP variants with the cell-adhesive (RGDS) to non-adhesive (RDGS) sequences within any stiffness regime. By manipulating these two variables, we can produce gels that have a spectrum of mechanical properties and ligand concentrations (Figure 3).
To encapsulate cells within 3D ELP hydrogels, the desired number of cells are suspended in the medium and centrifuged to produce a cell pellet (Figure 4A). The medium is aspirated from the tube, and the cells are re-suspended uniformly in the ELP solution of the desired concentration. Next, THPC solution is added to the cell/ELP suspension and pipetted thoroughly to form a homogenous mixture. This solution is quickly transferred to sterile silicone molds within a 24-well plate using a pipette and allowed to crosslink at room temperature and 37 °C for 15 min each (Figure 4B). Finally, the medium is added to the well plate and incubated at 37 °C in the experiment.
Live/dead staining can be used to assess cell viability and successful cell encapsulation within ELP hydrogels. As illustrated in Figure 5, adult murine neural progenitor cells (NPCs) show high cell viability over 7 days within a 3% (w/v) ELP hydrogel.
3D ELP hydrogels have been previously shown to support NPC stem maintenance measured through the expression of canonical NPC protein markers SRY (sex determining region Y)-box 2 (Sox2) and nestin9. NPCs encapsulated in 3% (w/v) ELP hydrogels with low THPC crosslinking show high expression of nuclear-localized Sox2 and cytoplasmic nestin filaments via immunostaining and confocal imaging (Figure 6).
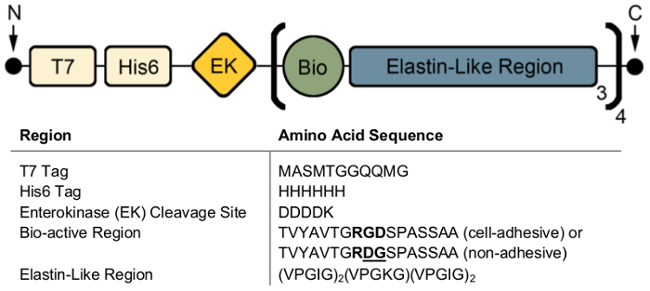
Figure 1: A schematic representation of the ELP and corresponding amino acid sequences. The ELP used in this study contains a T7 and His6 tag for antibody-based imaging, a bioactive region for introduction of cell-adhesive domains, and an elastin-like region that confers elastic mechanical properties and allows for chemical crosslinking. Please click here to view a larger version of this figure.
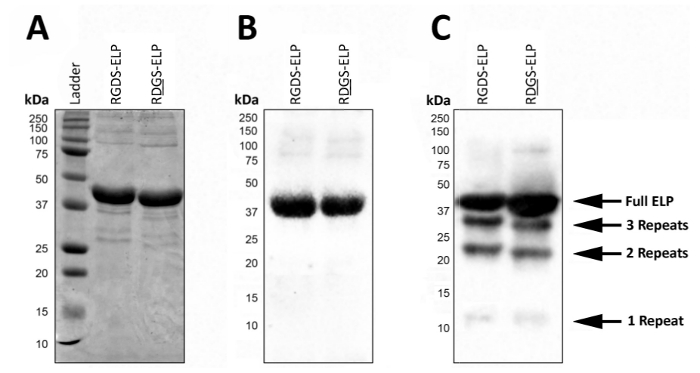
Figure 2: Target protein expression can be validated with SDS-PAGE and Western blot to confirm the molecular weight and identity of the final lyophilized product. Pure full-length ELP runs at a molecular weight of 37 kDa as reported by both SDS-PAGE (A) and Western blot using the T7 (MASMTGGQQMG) or histidine 6 (HHHHHH) tag (B). (C). Impure batches of ELP due to the deviations in the ELP expression protocol can lead to the expression of ELPs with lower molecular weights. Please click here to view a larger version of this figure.
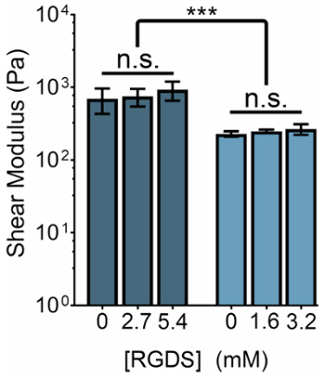
Figure 3: RGDS ligand content can be independently tuned from mechanical properties within ELP hydrogels. 5% (w/v) and 3% (w/v) ELP hydrogels have shear moduli of ~800 Pa and ~400 Pa, respectively. Hydrogels with a 1:1 ratio of THPC reactive groups:ELP primary amines were crosslinked at room temperature for 15 min, heated to 37 °C, and allowed to equilibrate for 5 min prior to measurement. Data are mean ± s.d., ***p <0.001. Please click here to view a larger version of this figure.
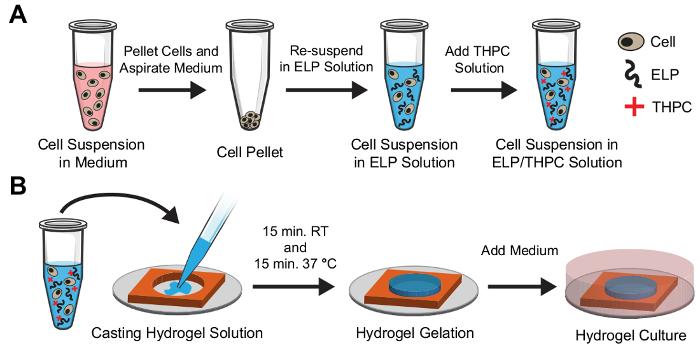
Figure 4: Schematic of cell encapsulation in ELP hydrogels. (A). Cells are initially dissociated into a single-cell suspension in the medium and pelleted using a centrifuge. The medium is aspirated from the tube, and the cells are re-suspended in ELP solution at the desired concentration and mixed well. Finally, the THPC crosslinking solution is added and mixed well. (B). Immediately following the addition of THPC, the solution is cast into a silicone mold with a pipette. The solution is allowed to crosslink at room temperature for 15 min followed by a second 15 min incubation at 37 °C. The medium is then added to the culture well for the duration of the experiment. Please click here to view a larger version of this figure.

Figure 5: Neural progenitor cells maintain high viability in ELP hydrogels. Representative image of neural progenitor cells encapsulated in 3% (w/v) ELP hydrogels with 1:1 crosslinking (THPC reactive groups:ELP primary amines) after 7 days in culture. Green: live staining (calcein-AM); Red: dead staining (ethidium homodimer). Scale Bar = 100 µm.
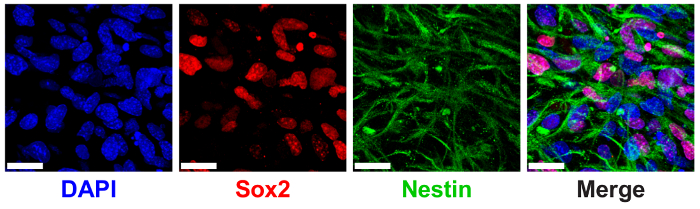
Figure 6: 3D ELP hydrogels support neural progenitor cell stem maker expression. Immunofluorescence image of neural progenitor cells expressing Sox2 and nestin proteins after 7 days of culture in ELP hydrogels. Images show cells encapsulated in 3% (w/v) ELP gels with 0.5:1 crosslinking (THPC reactive groups:ELP primary amines). Blue: DAPI (nuclei); Red: Sox2; Green: nestin. Scale Bar = 25 µm. Please click here to view a larger version of this figure.
Discussion
Recombinant protein expression and purification is a powerful tool to synthesize biomaterials with high reproducibility. Owing largely to the advent of commercialized molecular cloning, custom recombinant plasmids can be purchased from several suppliers, which significantly reduces the time to work with materials like ELPs. Similarly, plasmids can be requested directly from the originating lab when the original work was supported by a federal contract and the future work will be for non-profit use. The full ELP amino acid sequence has been previously published for several ELP variants31. However, the process from expression to eventual purification of recombinant proteins involves a number of critical steps that can commonly lead to reduced yields or a lower quality product. Some of the most common issues for ELP preparation arise in one of the following: (1) quality of stored bacterial stocks, (2) the first freeze-thaw cycle to disrupt the bacterial membrane, and (3) protein purification through thermal cycling.
A major difference between protein expression and other non-biological means of producing materials is that we are leveraging the biological machinery of recombinant hosts to synthesize the polymers. Subsequently, this technique comes with a unique limitation: cellular death or damage. Cell death most commonly manifests itself as a reduced number of bacterial colonies after streaking a plate or abnormally small colonies that grow relatively slowly. Bacterial stocks, if maintained carefully, can remain stable for years; however, successive freeze-thaw cycles due to repeated use or freezer failure can reduce cell viability or lead to DNA damage. Typical BL21 bacteria stocks use between 10% and 40% glycerol by volume mixed with suspended cells. The purpose of the glycerol is to reduce membrane damage from nucleating ice crystals during freezing. Therefore, using low concentrations (<10%) can lead to a compromised membrane, while higher concentrations (>40%) can suppress the freezing point sufficiently to where the stock never freezes leading to cell death. However, even within optimal glycerol levels, bacterial stocks should not be allowed to fully thaw as a combination of membrane damage from re-freezing and cytotoxic effects from the glycerol can lead to reduced stock viability and DNA damage. Therefore, if it is observed that a bacterial stock results in a low colony count or that the cells are dividing at a consistently slow rate (manifested as a slow OD600 ramp rate during expression), re-transforming the plasmid and making a new stock is a simple first approach to troubleshoot this problem. With this in mind, to ensure the long-term upkeep of bacterial stocks and integrity of DNA, it is best to store the copies of your plasmid as purified DNA frozen in water and not within cells. Storing DNA in this way will ensure that in unforeseen events such as a failed stock or freezer failure, a reliable source of the original DNA can be used for transformation.
Another critical step in ELP fabrication is the purification of the target protein from the expression host. Protein extraction from E. coli is achieved by breaking the cell wall using nucleating ice crystals that form throughout the suspended cell lysate upon freezing, which is further compounded with successive freeze-thaw cycles. Alternative methods for rupturing the cell wall can be utilized such as sonication or a press. In particular, consecutive freeze-thawing of the lysate is advantageous as it only requires a freezer and no other specialty equipment. However, this procedure nonspecifically releases DNA, RNA, and protein contaminants, in addition to proteases that have the potential to degrade the target protein. Therefore, to avoid contamination and reduced yield, deoxyribonuclease I (DNase) and phenylmethanesulfonyl fluoride (PMSF) are added to the cell lysate to degrade the DNA and inhibit proteases, respectively. The presence of DNA prior to the addition of DNase can be observed visually as a 'stringy' appearance throughout the re-suspended cell lysate following the first thaw. DNase actively degrades this DNA and thus reduces the viscosity of the cell lysate making it easier to purify via centrifugation. Optimal break down of DNA can be visually confirmed by ensuring that the cell lysate appears to be entirely liquid and that the stringy appearance is no longer visible. We have observed in practice that the addition of ~0.1 mg of DNase per mL cell lysate is sufficient to achieve necessary degradation. However, if the presence of DNA is still observed, more DNase can be added followed by an additional two to three hours of agitation. A similar issue can also arise if DNase is added prematurely before any of the lysate has had the potential to sufficiently thaw. In this case, the colder temperatures can limit the efficiency of DNA degradation due to the premature inactivation of DNase. To avoid this issue, it is often best practice to allow the re-suspended pellet to thaw for approximately 8 hours prior to treatment with DNase. In addition, if low protein yields are reported and the breakdown of DNA has been sufficient, the addition of more PMSF to help further reduce potential protein degradation from proteases may be required.
Additional considerations for ensuring optimal expression of ELPs include a careful understanding of the benefits and limitations of a chosen antibiotic. Here, pET15b vectors containing an ampicillin resistance gene were used for protein expression. Functionally, the pET vector series allow for significant protein expression with as much as 50% of a bacterium's protein expression dedicated to the target protein following a successful induction32,33. However, ampicillin as a selection antibiotic comes with some limitations that may interfere with optimum expression. First, degradation of ampicillin in the presence of E. coli can occur rapidly due to the release of beta-lactamase. If a sufficient quantity of the ampicillin is degraded, the ampicillin-encoding plasmid (i.e. the ELP-encoding plasmid) may be lost entirely. As a result, when expressing ELPs for longer durations, protein expression levels should be carefully monitored at successive time points to ensure sufficient amount of the ELP-encoding gene remains to allow for desirable expression. Possible methods for troubleshooting the buildup of beta-lactamases include spinning down the starter culture and re-suspending the cells in antibiotic-free medium prior to inoculating the expression medium. This process effectively limits the transfer of antibiotic-degrading enzymes and ensures a greater portion of the cells contain the target-protein-encoding vector. Additionally, ampicillin has a limited shelf life of approximately two to three weeks. Therefore, culture plates for protein expression should be stored at 4 °C for a maximum of two weeks prior to use. Finally, to ensure the efficacy of ampicillin within the starter and expression media, the ampicillin stock solution should be produced fresh immediately before use, as long-term storage may lead to a less effective antibiotic.
The presence of an LCST allows for the simple purification of ELPs through thermal cycling. Specifically, at a higher temperature and in the presence of salts, entropic forces cause ELPs to become less soluble and subsequently form a polymer-rich coacervate phase. On the other hand, at lower temperatures, ELPs remain soluble and readily dissolve into the solution. Cycling between these two temperature regimes coupled with centrifugation steps to collect and discard the non-ELP-containing phase successively concentrates the protein and simultaneously reduces the existence of non-ELP contaminants.
However, there are a number of stages where ELPs can be lost in this purification process. First, prior to every cold spin, the protein-containing solution is alkalized to a pH of 9.0. This higher pH serves to deprotonate certain amino acids on the protein backbone, effectively leaving them in a charged state and further enhancing their solubility. Consequently, foregoing this step or not allowing sufficient time for protein dissolution can lead to a reduction in yield as non-solubilized proteins will be pelleted during centrifugation and discarded.
Similarly, target proteins can be lost during the hot spin procedure when the ELP is pelleted. Initially, NaCl is added to the protein-rich supernatant to reduce the solubility of the ELP. The salts work to shield electrostatic interactions between the protein and water molecules, causing the protein to separate from the aqueous phase. This effect is amplified by heating the solution, which, due to entropic effects, further breaks down the hydrous 'cage' surrounding ELPs and forces the aggregation of the proteins. At lower protein concentrations (i.e., the first thermal cycle), the addition of salts alone is often insufficient to cause this phase separation. However, as the concentration of protein increases (i.e., later thermal cycles), and there are less secondary contaminants to interact with the salts, the ELP will more readily precipitate. As a result, if salts are added too quickly, they may become physically trapped by aggregating proteins, which effectively reduces the salt concentration of the solution and limits further protein precipitation. Thus, the salt should be added in three small batches to ensure they have sufficient time to homogenously distribute through the solution. As a final note, variations to the ELP backbone, either through further modifications to the guest residue of the elastin-like region or changes to the bio-active region can significantly impact the LCST behavior. Consequently, to ensure optimal protein yields across protein variants, it is crucial to optimize the pH, salt concentration, and salt type (e.g., monovalent or divalent) for the cold and hot spins.
Running SDS-PAGE upon protocol completion is recommended as it can be used to easily determine if significant ELP loss occurs during any of the purification steps. Briefly, if ELP is detected in the supernatant following a hot spin, then the protein is not being effectively precipitated. Similarly, if ELPs are identified in a sample of solubilized pellet following a cold spin, then the protein is not being effectively dissolved.
ELP hydrogels offer many advantages over synthetic or naturally-derived materials. Specifically, the use of the amine-reactive crosslinker THPC affords a low-cost, simple, and tunable mechanism of protein crosslinking. However, there are distinct limitations within the crosslinking protocol that should be noted. THPC is oxygen sensitive, and if stored under improper conditions, it can quickly deteriorate in reaction efficiency. In addition, due to its reactivity with primary amines, THPC may react with surrounding proteins in media or those on the cell surface that are rich in amines. Therefore, when forming ELP hydrogels, it is recommended to avoid media contamination with the cell pellet to reduce possible exogenous protein cross-reactivity and thus, a reduction in crosslinking efficiency. Finally, this crosslinking mechanism precludes the bio-active region sequence to those containing no lysine residues and thus, limits potential integration of some cell-adhesive motifs (e.g., IKVAV34). To address these limitations, modifications to the ELP backbone with azide and bicyclononyne (BCN) reaction partners allows for bio-orthogonal crosslinking, as previously described27.
It should be noted that the ELP LCST behavior plays an important role in dictating hydrogel microstructure. At temperature regimes above the LCST, ELPs precipitate out of solution leading to the formation of protein-rich and protein-deficient phases that can influence matrix porosity and crosslinking efficiency of the matrix9. Because most cell culture experiments are conducted at physiologically relevant temperatures (~37 °C) above the ELP LCST, these effects should be considered. For the hydrogels to effectively crosslink and form an interconnected protein network, the primary amine from the lysine must be physically accessible to the THPC crosslinker. If the ELP aggregation occurs before reaching sufficient crosslinking, ELPs trapped within the protein-rich phase may be inaccessible and thus unable to participate in crosslinking. To address this limitation, our protocol requires an initial 15 min crosslinking period at room temperature, which allows for preliminary crosslinking of the hydrogel before the ELP undergoes its thermal phase transition. This room temperature incubation is followed by an additional 15 min incubation at 37 °C to finalize hydrogel crosslinking. This procedure is critical for sufficient crosslinking and robust, reproducible gelation of the ELP material.
In conclusion, recombinant protein hydrogels fabricated using ELP offer exceptional tunability of the protein sequence and therefore the 3D cell microenvironment. ELP polymers have been shown to be expressible in high yields, easily purified owing to their LCST behavior, and biocompatible in a wide variety of in vitro and in vivo systems. The use of E. coli as a recombinant host provides a simple and inexpensive procedure that gives rise to near perfect control of polymer molecular weight and functionality. In conjunction, this technique allows for robust tunability and reproducibility of the hydrogel platform allowing for the culture of a wide range of cell types in 3D. Finally, this ELP hydrogel platform is amenable to many downstream biochemical assays including qRT-PCR, Western blot, DNA extraction, and cell immunostaining9.
Declarações
The authors have nothing to disclose.
Acknowledgements
The authors thank T. Palmer and H. Babu (Stanford Neurosurgery) for providing murine NPCs. Vector art in Figure 4 was used and adapted from Servier Medical Art under Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/legalcode). Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152. N.A.S. acknowledges support from the National Institute of General Medical Sciences of the National Institutes of Health (32GM008412). C.M.M. acknowledges support from an NIH NRSA pre-doctoral fellowship (F31 EB020502) and the Siebel Scholars Program. S.C.H. acknowledges support from the National Institutes of Health (U19 AI116484 and R21 EB018407), National Science Foundation (DMR 1508006), and the California Institute for Regenerative Medicine (RT3-07948). This research received funding from the Alliance for Regenerative Rehabilitation Research & Training (AR3T), which is supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institute of Neurological Disorders and Stroke (NINDS), and National Institute of Biomedical Imaging and Bioengineering (NIBIB) of the National Institutes of Health under Award Number P2CHD086843. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institutes of Health.
Materials
| Elastin-Like Protein Expression and Purification | |||
| 10 cm Petri Dishes | Thermo Fisher Scientific | FB0875713 | |
| 70% Ethanol | RICCA Chemical | 2546.70-1 | |
| Ammonium Sulfate | Sigma-Aldrich | A3920-500G | |
| Ampicillin | Thermo Fisher Scientific | BP1760-25G | |
| Bacto Agar | Thermo Fisher Scientific | 9002-18-0 | |
| BL21(DE3)pLysS Competent Cells | Invitrogen | C606003 | |
| Chloramphenicol | Amresco | 0230-100G | |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | DN25 | |
| EDTA disodium salt, dihydrate | Thermo Fisher Scientific | O2793-500 | |
| Glycerol | Thermo Fisher Scientific | BP229-4 | |
| Isopropanol | Thermo Fisher Scientific | A451-4 | |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Thermo Fisher Scientific | BP1755-10G | |
| Luria Broth | EMD Millipore | 1.10285.5007 | |
| Parafilm | VWR | 52858-000 | |
| Phenylmethanesulfonyl fluoride (PMSF) | MP Biomedicals | 195381 | |
| Sodium Chloride | Thermo Fisher Scientific | BP358-212 | |
| Sodium Hydroxide | Sigma-Aldrich | S 8045-1KG | |
| Syringe Filter Unit (0.22 μm) | Millipore | SLGP033RB | |
| Terrific Broth | Millipore | 71754-4 | |
| Tris Base | Thermo Fisher Scientific | BP152-1 | |
| Cell Encapsulation in 3D ELP Hydrogels | |||
| 0.22 μm syringe filters | Millipore | SLGV004SL | |
| 0.5 mm thick silicone sheet | Electron Microscopy Science | 70338-05 | |
| 24-well tissue culture plates | Corning | 353047 | |
| Disposable Biopsy Punch (2 mm) | Integra Miltex | 33-31 | |
| Disposable Biopsy Punch (4 mm) | Integra Miltex | 33-34 | |
| Disposable Biopsy Punch (5 mm) | Integra Miltex | 33-35 | |
| Dulbecco’s phosphate buffered saline (DPBS) | Corning | 21-031-CM | |
| No. 1 12 mm glass coverslips | Thermo Fisher Scientific | 12-545-80 | |
| Tetrakis(hydroxymethyl)phosphonium chloride (THPC) | Sigma-Aldrich | 404861-100ML | |
| 0.5% Tryspin/EDTA | Thermo Fisher | 15400054 | |
| Immunocytochemistry of Cells in 3D ELP Hydrogels | |||
| 16% (w/v) Paraformaldehyde (PFA) | Electron Microscopy Sciences | 15701 | |
| Bovine Serum Albumin (BSA) | Roche | 3116956001 | |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Molecular Probes | D1306 | |
| Donkey Serum | Lampire Biological Labs | 7332100 | |
| Goat anti-mouse Secondary Antibody (AF488) | Molecular Probes | A-11017 | |
| Goat anti-rabbit Secondary Antibody (AF546) | Molecular Probes | A-11071 | |
| Goat Serum | Gibco | 16210-072 | |
| Mouse Nestin Primary Antibody | BD Pharmingen | 556309 | |
| Mouse Sox2 Primary Antibody | Cell Signaling Technology | 23064S | |
| Nail Polish | Electron Microscopy Sciences | 72180 | |
| Triton X-100 | Sigma-Aldrich | X100-100ML | |
| Vectashield Hardset Mounting Medium | Vector Labs | H-1400 |
Referências
- Cukierman, E., Pankov, R., Stevens, D. R., Yamada, K. M. Taking Cell-Matrix Adhesions to the Third Dimension. Science. 294 (5547), 1708-1712 (2001).
- Birgersdotter, A., Sandberg, R., Ernberg, I. Gene expression perturbation in vitro-A growing case for three-dimensional (3D) culture systems. Seminars in Cancer Biology. 15 (5), 405-412 (2005).
- Gómez-Lechón, M. J., et al. Long-term expression of differentiated functions in hepatocytes cultured in three-dimensional collagen matrix. Journal of Cellular Physiology. 177 (4), 553-562 (1998).
- Baker, B. M., Chen, C. S. Deconstructing the third dimension – how 3D culture microenvironments alter cellular cues. Journal of Cell Science. 125 (13), 3015-3024 (2012).
- Pampaloni, F., Reynaud, E. G., Stelzer, E. H. K. The third dimension bridges the gap between cell culture and live tissue. Nature Reviews Molecular Cell Biology. 8 (10), 839-845 (2007).
- Justice, B. A., Badr, N. A., Felder, R. A. 3D cell culture opens new dimensions in cell-based assays. Drug Discovery Today. 14 (1-2), 102-107 (2009).
- Caliari, S. R., Burdick, J. A. A practical guide to hydrogels for cell culture. Nature Methods. 13 (5), 405-414 (2016).
- Tibbitt, M. W., Anseth, K. S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnology and Bioengineering. 103 (4), 655-663 (2009).
- Madl, C. M., et al. Maintenance of neural progenitor cell stemness in 3D hydrogels requires matrix remodelling. Nature Materials. 16 (12), 1233-1242 (2017).
- Discher, D. E., Janmey, P., Wang, Y. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science. 310 (5751), 1139-1143 (2005).
- Sun, Y., Villa-Diaz, L. G., Lam, R. H. W., Chen, W., Krebsbach, P. H., Fu, J. Mechanics Regulates Fate Decisions of Human Embryonic Stem Cells. PLoS ONE. 7 (5), e37178 (2012).
- Ehrbar, M., et al. Elucidating the Role of Matrix Stiffness in 3D Cell Migration and Remodeling. Biophysical Journal. 100 (2), 284-293 (2011).
- Rowlands, A. S., George, P. A., Cooper-White, J. J. Directing osteogenic and myogenic differentiation of MSCs: interplay of stiffness and adhesive ligand presentation. American Journal of Physiology – Cell Physiology. 295 (4), 1037-1044 (2008).
- Lampe, K. J., Antaris, A. L., Heilshorn, S. C. Design of three-dimensional engineered protein hydrogels for tailored control of neurite growth. Acta Biomaterialia. 9 (3), 5590-5599 (2013).
- Kilian, K. A., Mrksich, M. Directing Stem Cell Fate by Controlling the Affinity and Density of Ligand-Receptor Interactions at the Biomaterials Interface. Angewandte Chemie International Edition. 51 (20), 4891-4895 (2012).
- Tse, J. R., Engler, A. J. Preparation of Hydrogel Substrates with Tunable Mechanical Properties. Current Protocols in Cell Biology. , 10.16.1-10.16.16 (2010).
- Hughes, C. S., Postovit, L. M., Lajoie, G. A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics. 10 (9), 1886-1890 (2010).
- DiMarco, R. L., Heilshorn, S. C. Multifunctional Materials through Modular Protein Engineering. Advanced Materials. 24 (29), 3923-3940 (2012).
- Meyer, D. E., Chilkoti, A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nature Biotechnology. 17 (11), 1112-1115 (1999).
- Aladini, F., Araman, C., Becker, C. F. W. Chemical synthesis and characterization of elastin-like polypeptides (ELPs) with variable guest residues. Journal of Peptide Science. 22 (5), 334-342 (2016).
- McMillan, R. A., Caran, K. L., Apkarian, R. P., Conticello, V. P. High-Resolution Topographic Imaging of Environmentally Responsive, Elastin-Mimetic Hydrogels. Macromolecules. 32 (26), 9067-9070 (1999).
- McMillan, R. A., Conticello, V. P. Synthesis and Characterization of Elastin-Mimetic Protein Gels Derived from a Well-Defined Polypeptide Precursor. Macromolecules. 33 (13), 4809-4821 (2000).
- Chung, C., Lampe, K. J., Heilshorn, S. C. Tetrakis(hydroxymethyl) Phosphonium Chloride as a Covalent Cross-Linking Agent for Cell Encapsulation within Protein-Based Hydrogels. Biomacromolecules. 13 (12), 3912-3916 (2012).
- Romano, N. H., Madl, C. M., Heilshorn, S. C. Matrix RGD ligand density and L1CAM-mediated Schwann cell interactions synergistically enhance neurite outgrowth. Acta Biomaterialia. 11, 48-57 (2015).
- Shah, M., Hsueh, P. Y., Sun, G., Chang, H. Y., Janib, S. M., MacKay, J. A. Biodegradation of elastin-like polypeptide nanoparticles. Protein Science. 21 (6), 743-750 (2012).
- Nettles, D. L., Chilkoti, A., Setton, L. A. Applications of elastin-like polypeptides in tissue engineering. Advanced Drug Delivery Reviews. 62 (15), 1479-1485 (2010).
- Madl, C. M., Heilshorn, S. C. Tyrosine-Selective Functionalization for Bio-Orthogonal Cross-Linking of Engineered Protein Hydrogels. Bioconjugate Chemistry. 28 (3), 724-730 (2017).
- Zhu, D., Wang, H., Trinh, P., Heilshorn, S. C., Yang, F. Elastin-like protein-hyaluronic acid (ELP-HA) hydrogels with decoupled mechanical and biochemical cues for cartilage regeneration. Biomaterials. , 132-140 (2017).
- Madl, C. M., Katz, L. M., Heilshorn, S. C. Bio-Orthogonally Crosslinked, Engineered Protein Hydrogels with Tunable Mechanics and Biochemistry for Cell Encapsulation. Advanced Functional Materials. 26 (21), 3612-3620 (2016).
- Cai, L., Dinh, C. B., Heilshorn, S. C. One-pot synthesis of elastin-like polypeptide hydrogels with grafted VEGF-mimetic peptides. Biomater Sci. 2 (5), 757-765 (2014).
- Straley, K. S., Heilshorn, S. C. Independent tuning of multiple biomaterial properties using protein engineering. Soft Matter. 5 (1), 114-124 (2009).
- Baneyx, F. Recombinant protein expression in Escherichia coli. Current Opinion in Biotechnology. 10 (5), 411-421 (1999).
- Graumann, K., Premstaller, A. Manufacturing of recombinant therapeutic proteins in microbial systems. Biotechnology Journal. 1 (2), 164-186 (2006).
- Tashiro, K., et al. A Synthetic Peptide Containing the IKVAV Sequence from the A Chain of Laminin Mediates Cell Attachment, Migration, and Neurite Outgrowth. Journal of Biological Chemistry. 264 (27), 16174-16182 (1989).
