The workflow for the entire protocol is depicted in Figure 1. It consists of two variations defined by the number of cells sorted: either limiting dilution or as single cells, as described in the text. Examples of primer-probe qualification analyses on 2-fold serial RNA dilutions are shown in Figure 2. The gating strategy to identify potential SIV+ cells is shown in Figure 3. A successful, suboptimal and failed quality control qPCR for the housekeeping gene GAPDH on FACS-sorted cells in limiting dilution are shown in Figure 4. Single-cell quantitative expression of four SIV RNA species and rhesus macaque genes that were differentially expressed in infected cells are shown in Figure 5. Representative bivariate, histogram, and scatterplots depict the surface protein expression profile of SIV RNA+ CD4+ T cells measured by flow cytometry in Figure 6. The trivariate bubble plot (Figure 7) displays the relationship between surface CD3 or CD4 protein, CD3 or CD4 mRNA, and quantitative viral gene (tat/rev) expression in single cells.
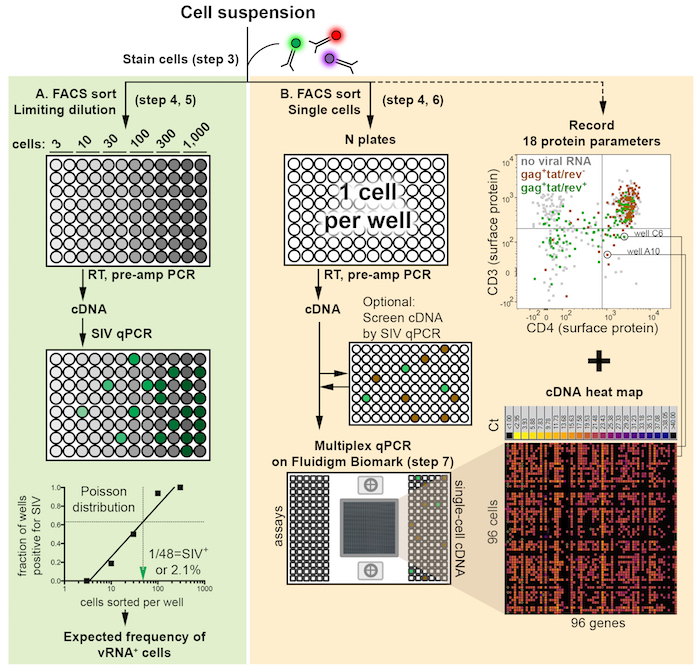
Figure 1: Schematic of experimental workflow illustrates three major components: flow cytometric sort, reverse transcription plus PCR-based pre-amplification of cDNA (RT, pre-amp), and qPCR. The sort can be performed as either a limiting dilution (A, green backdrop) or as single cells (B, orange backdrop). Immediately following the FACS sort, cells are lysed and RNA is reverse transcribed into cDNA and pre-amplified (RT, pre-amp PCR) to prepare qPCR template. Limiting dilution sorts determine the frequency of cells positive for viral RNA using Poisson distribution statistics as well as experimental efficiency and sample recovery. The green arrow head indicates the estimated number of cells sorted per well containing one cell positive for a viral gene (corresponding to a 63.2% probability of such wells being gene positive), which is converted into a cell frequency. Frequency estimates may be used to inform the number of single cells collected in a subsequent sort (B). Indexed single-cell FACS sorting deposits one cell per well and generates data files for each cell annotated by well position within the 96-well plate. Single-cell qPCR is performed in multiplex for 96 genes simultaneously. Combining the surface protein (FACS) and mRNA expression allows for profiling of individual cells (right column). An optional qPCR for a viral gene may be performed between the pre-amplification PCR and multiplexed qPCR (B, middle) to screen single cells or pools of single-cell cDNA to down-select viral RNA+ cells or pools for multiplex qPCR analysis. The heat map illustrates gene expression (Ct value) for 96 assays (columns) and 96 single cells (rows). Please click here to view a larger version of this figure.
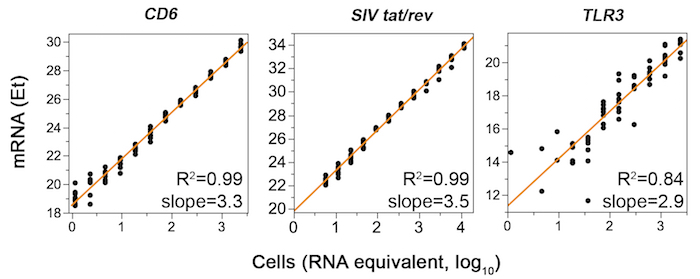
Figure 2: Representative qPCR data from primer qualification experiments are shown for successful (left and middle) and failed (right) commercially available assays (CD6, SIV tat/rev, and TLR3, respectively). For CD6 and TLR3, RNA was extracted from 106 FACS-sorted rhesus macaque PBMC CD4+ T-cells using a commercial kit. Eight replicates of a twelve-point RNA two-fold dilution series (0.023-48 ng RNA, corresponding to 1.2–2,400 cell equivalents assuming 20 pg RNA per CD4+ T-cell) were subjected to RT-preamp and qPCR. For SIV tat/rev, RNA was extracted from rhesus macaque PBMCs infected in vitro with SIVmac239. RNA dilutions were prepared spanning RNA equivalents of 6–12,000 cells. Et (40-Ct) values, which increase with gene expression, are plotted versus estimated cell numbers. Dilution series exhibiting R2 >0.97 and slope of 3.32 ±0.3 indicate successful primer qualification. Please click here to view a larger version of this figure.
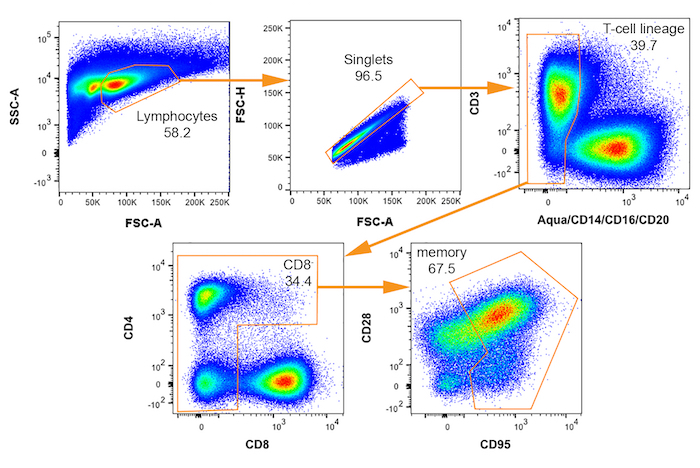
Figure 3: Bivariate FACS plots with gating scheme for isolation of rhesus macaque cells potentially infected by SIV. Sequential gates for selecting memory (CD95+) CD4+ T cells are shown from upper left to lower right with each population name indicated. Percent of parent plot that falls within each gate is indicated. Please click here to view a larger version of this figure.
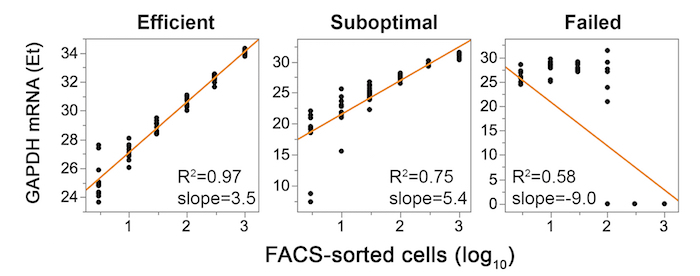
Figure 4: Representative experimental validation of qPCR performed on FACS-sorted cells in limiting dilution using GAPDH housekeeping gene for three independent experiments: successful (left), suboptimal (middle), and failed (right). Replicates of 10-100 cells per well should span no more than 2 Ets, and 300–1,000 cell wells within 1 Et. The linear regression slope should be 3.32 ± 0.3, R2 >0.95. Failure to achieve these specifications indicates technical difficulties in steps 1, 2, 3, or a combination thereof. Please click here to view a larger version of this figure.
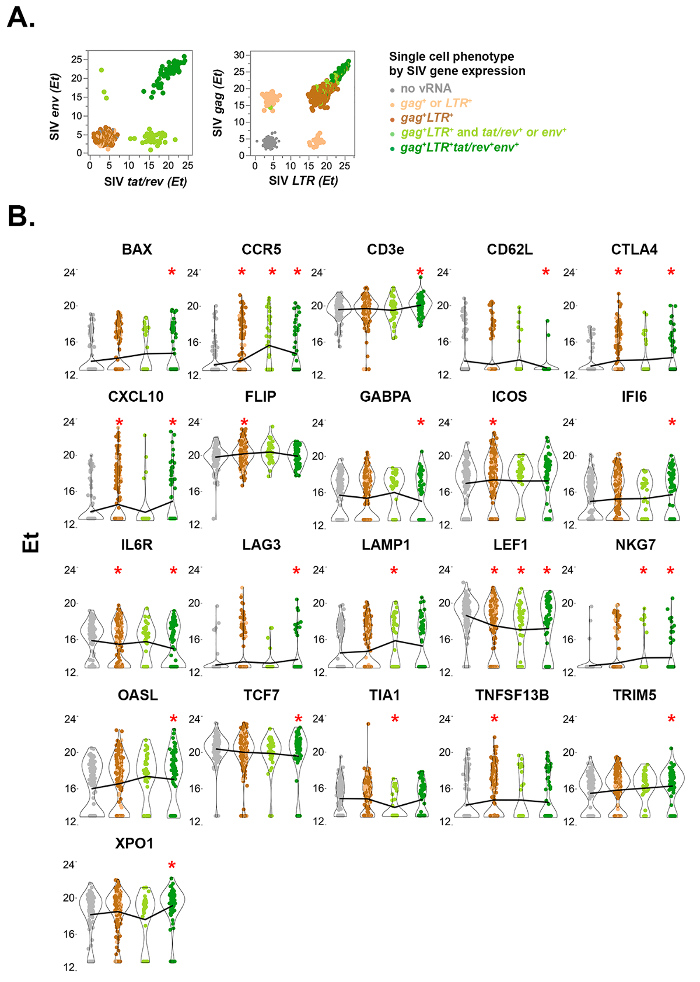
Figure 5: Single-cell quantitative viral and host gene expression in FACS-sorted rhesus macaque CD4+ T cells from mesenteric lymph node 10 days post-SIVmac251 infection. (A) Viral gene expression bivariate plots of multiply-spliced (tat/rev) and singly-spliced (env) SIV mRNA by single cells (left). Tat/rev+env– cells (light green cells along x-axis) express fewer copies of tat/rev RNA than the env+ cells (dark green), consistent with an early stage of infection and prior to Rev protein-mediated stabilization and nuclear export of partially spliced vRNAs such as env. Cells that do not express spliced viral RNA are depicted in grey (no viral RNA), brown (gag+ and LTR+), or tan (either gag+ or LTR+). Unspliced (gag+) and total (LTR+) SIV mRNA expression is shown for the same cells (right). High abundance of unspliced gag RNA in tat/rev+env+ cells is consistent with late stage productive infection during which abundant genomic RNA is expressed for packaging into budding virions. (B) Violin plots of rhesus macaque genes differentially expressed in at least one subset of SIV-infected cells compared to uninfected cells (gray). Statistical analysis was performed as described previously20,22,23,24. Asterisk indicates false discovery rate < 10% in combined likelihood ratio test comparisons relative to uninfected cells. Line connects mean values across cell groups for each gene. This figure has been modified from Bolton et al.20 Please click here to view a larger version of this figure.
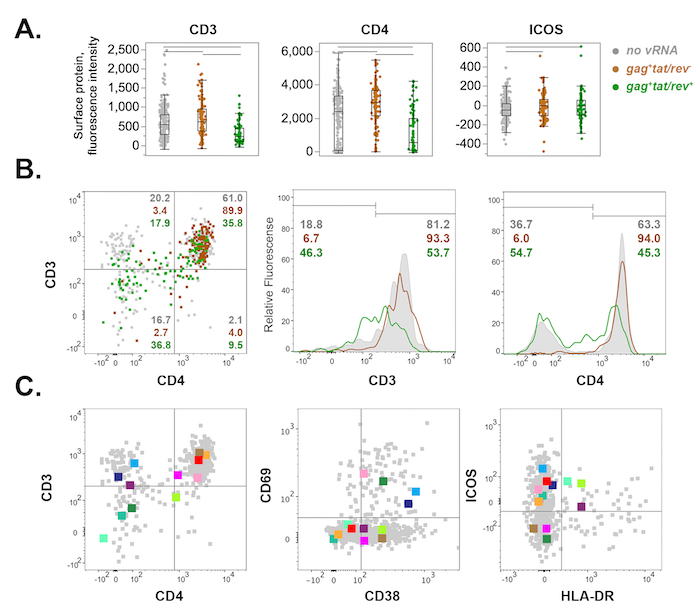
Figure 6: Representative host surface protein expression profiles of FACS-sorted cells from rhesus macaque mesenteric lymph node 10 days post-SIVmac251 infection. (A) Scatterplot display of CD3, CD4, and ICOS surface protein expression in uninfected (grey), gag+tat/rev– (brown), and gag+tat/rev+ cells (green). Fluorescence intensity is plotted for each cell (dot). Outlier box plots depict the interquartile range (IQR) and median (box), the furthest points within 1.5 x IQR from the box (whiskers), and potential outliers (disconnected points). Horizontal bars at the top indicate significant differences (p <0.05, nonparametric Wilcoxon rank test). (B) Bivariate and histogram display of surface CD3 and CD4 protein expression for cells shown in (A). Dot plot (left) indicates percentages of each cell population within a quadrant. CD3 and CD4 histograms (middle, right) depict surface protein downregulation among gag+tat/rev+ cells relative to uninfected and gag+tat/rev– cells. (C) Twelve representative tat/rev+ single cells from (A–B) are shown across three bivariate plots for surface expression of CD3/CD4 (left), CD69/CD38 (middle), and ICOS/HLA-DR (right). Please click here to view a larger version of this figure.
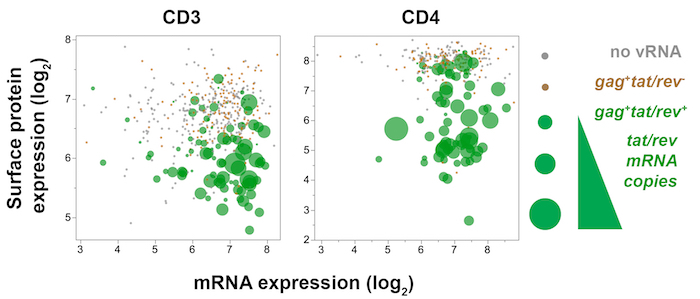
Figure 7: Trivariate plot displaying single-cell viral gene (tat/rev), host gene (CD3 or CD4), and host surface protein (CD3 or CD4) expression in SIV-infected memory CD4+ T cells from mesenteric lymph node 10 days post-SIVmac251 infection. CD4 protein expression (fluorescence) is plotted against CD4 mRNA (qPCR), while the amount of tat/rev expressed by each cell is reflected by dot size. In tat/rev+ cells (green), decreased surface CD4 and CD3 protein expression with sustained CD4 and CD3 transcripts, respectively, indicate that the surface protein expression is modulated downstream of gene expression. Please click here to view a larger version of this figure.
Table 1: Primers and probes used for the detection of SIV nucleic acids. When two sequences are indicated for a primer or probe, equimolar amounts of both sequences were used. Please click here to download this file.
Table 2: A 96-gene panel used for quantitation of amplicons on Biomark instrument. Four SIV assays are indicated with blue background. Please click here to download this file.
Table 3: Reaction mix used for reverse transcription and pre-amplification. Please click here to download this file.
Table 4: qPCR reaction mix used for real-time PCR performed on a Quant Studio 6 instrument. Please click here to download this file.
Supplemental Coding File 1. Instructions for qualifying gene expression assays. Please click here to download this file.
Supplemental Coding File 2: Sample Map template in JMP. Please click here to download this file.
Supplemental Coding File 3: Probe Map template in JMP. Please click here to download this file.
Supplemental Coding File 4: Primer Analysis script for JMP. Please click here to download this file.
Supplemental Coding File 5. Piecewise analysis script for JMP. Please click here to download this file.
