Examples provided below illustrate results obtained from IG gene sequencing analysis following the steps detailed above. Although DNA and/or RNA extraction are routine procedures for most clinical/research laboratories, a first critical step involves checking the quality/quantity of gDNA and/or RNA using a spectrophotometer or other suitable technology with high sensitivity. The next crucial step involves PCR amplification of the clonotypic IGHV-IGHD-IGHJ gene rearrangement. As mentioned above, only the use of IGHV leader primers enables amplification of the full length of the rearranged IGHV gene sequence, thus permitting the true SHM load to be determined; therefore, IGHV leader primers are the recommended primer choice (Figure 2). In rare instances where IGHV leader primers cannot amplify the clonotypic IG rearrangement, IGHV FR1 primers may be used. As evidenced in Figure 3, several PCR products/bands may be observed, especially when gDNA is the starting material; these bands should be excised and sequenced seperately. If both IGHV leader and IGHV FR1 primers fail to yield results, the analysis should be repeated using a new patient sample (when possible). The last checkpoint prior to sequencing is determining the clonality of the sample using fragment analysis (Figure 4).
Sequences obtained should be analyzed using the IMGT/V-QUEST tool30. User-selected parameters include species, receptor type or locus, search for insertion, and deletion option, etc. and are listed together with the number of sequences analyzed. Each FASTA sequence is displayed and the V-DOMAIN analyzed by IMGT/V-QUEST is highlighted in green. Additionally, if the input sequence was in the opposite orientation (antisense), the program automatically provides the complementary reverse sequence and states this above the FASTA sequence.
The Result summary table is provided directly below the FASTA sequence, at the top of the 'Detailed View' results page, and contains information important for the clinical reporting of IG gene sequence analysis in CLL. Each row of this table is explained below.
Result summary. The first row in the results table states the functionality of the input sequence: an IG rearranged sequence can be either productive or unproductive (Figure 5A & 5B). An IG gene rearrangement is unproductive if stop codons are present within the V-D-J-REGION (for VH) or within the V-J-REGION (for VL) and/or if the rearrangement is out-of-frame due to insertions/deletions. A third output is provided when the functionality cannot be determined, i.e., if the IGHV-IGHD-IGHJ junction could not be identified; in this instance, the result summary would read as 'No rearrangement found' or 'No junction found'. It is important to note that only productive and, thus, functional rearrangements should be analyzed further. If the sole amplified IG rearrangement is not functional (unproductive or 'no rearrangement found'), the PCR amplification process should be repeated. If the result remains negative a new patient sample should be obtained.
V-GENE and allele. The 'V-GENE and allele' row indicates the closest germline IGHV gene and allele with its alignment score and the percent identity. If several gene(s) and alleles give the same high score, all are listed. The percent identity between the submitted sequence and the closest IGHV gene and allele is of particular importance since it determines the SHM status. This value is calculated from the first nucleotide of the V-REGION to the 3' end of the V-REGION excluding the CDR3. If IGHV FR1 primers are used, the sequence corresponding to that of the primer should always be removed to ensure as accurate a result as possible. It should be noted that a low identity percentage (<85%) may result from an insertion or deletion (discussed further below) and, should this be the case a 'Warning' note will appear in the 'Result summary' row to alert the user.
J-GENE and allele. As described for the V-gene and allele above, the 'J-GENE and allele' row indicates the closest germline IGHJ gene and allele with its alignment score and the percent identity. However, as the IGHJ gene is rather short and its 5' end may have been trimmed due to exonuclease activity, alternative choices are also searched by the tool, based on the highest number of consecutive identical nucleotides.
D-GENE and allele by IMGT/JunctionAnalysis. The 'D-GENE and allele by IMGT/JunctionAnalysis' indicates the closest germline IGHD gene and allele with the D-REGION reading frame identified by the tool in the user sequence. IMGT/JunctionAnalysis31 provides a detailed and accurate analysis of the junction and has been integrated into the IMGT/V-QUEST tool interface. This tool manages all aspects of difficulty linked to IGHD gene and allele identification i.e., (i) the short length of the D-REGION; (ii) use of 2 or even 3 reading frames; (iii) exonuclease trimming at both ends of the gene; and, (iv) the presence of mutations.
FR-IMGT lengths, CDR-IMGT lengths and AA JUNCTION. This row provides the length of the IGHV FRs and CDRs shown within brackets and separated by dots and the amino acid (AA) junction. The AA sequence of the junction illustrates (i) an in- or out-of-frame rearrangement (out-of-frame positions are indicated by the '#' sign), (ii) the presence or absence of stop codons (a stop codon is shown by an asterisk '*'), and (iii) the presence or absence of the anchors of the VH CDR3-IMGT: C (2nd-CYS 104) and W (J-TRP 118).
Somatic hypermutations predominantly consist of single nucleotide changes, however, small insertions and deletions within the V-region can be observed, albeit at a much lower frequency33. When using the default IMGT/V-QUEST parameters, insertions and deletions are not automatically detected; however, strong indications that a sequence may carry such alterations, i.e., a low percent identity and/or different IGHV CDR1-IMGT and CDR2-IMGT lengths between the submitted user sequence and the closest germline gene and allele, are detected by the tool and a warning alert appears below the Results summary table (Figure 5C).
IMGT provides an option called 'Search for insertions and deletions' in the 'Advanced parameters' section located below the 'Display results' section. In cases where relevant warnings appear, it is advisable to use this functionality. When insertions and deletions are detected, comprehensive details of the finding are provided in the 'Result summary' section including (i) where the insertion or deletion is located i.e., VH FR-IMGT or CDR-IMGT; (ii) the number of inserted/deleted nucleotides; (iii) the sequence (only for insertions); (iv) whether a frameshift has occurred; (v) the V-REGION codon from which the insertion or deletion starts; and (vi) the affected nucleotide position in the submitted sequence (Figure 5D). For insertions, the tool removes the insertion(s) from the submitted user sequence and then re-analyzes the sequence using the standard IMGT/V-QUEST parameters. If deletions are detected, the tool adds gaps, represented by dots, to replace the deleted nucleotides before repeating the analysis.
As for every diagnostic or prognostic test performed in clinical laboratories, stringent standards and a high level of reproducibility are of utmost importance. The IMGT/V-QUEST output provides much of the information necessary for reporting the results of IG gene sequence analysis in CLL, i.e., (i) the IGHV, IGHD and IGHJ genes and alleles utilised; (ii) the functionality of the clonotypic IGHV-IGHD-IGHJ gene rearrangement i.e., whether the rearrangement is productive or unproductive; and (iii) the percent identity of the rearranged IGHV gene and allele in comparison to its closest germline IGHV gene and allele. For comprehensive details regarding the clinical reporting of IG genes in CLL, the interpretation of problematic or technically challenging cases and recommendations for the accurate and robust determination of IGHV gene SHM status in CLL, the reader is directed to recently updated ERIC guidelines and reports27,34.
Finally, owing to our growing knowledge concerning BcR IG stereotypy in CLL, an additional recommended requirement for the clinical reporting of IG gene rearrangements in CLL relates to the assignment of cases to major stereotyped subsets. It is now recommended to state in the clinical report whether the analyzed productive IGHV gene rearrangement can be assigned to one of the major stereotyped subsets, namely subsets #1, #2, #4 or #812,27. Assignment to major CLL stereotyped subsets should be performed with the use of the ARResT/AssignSubsets tool (Figure 6)32.
| 5’ IGHV FR1 primers | Primer sequence | Quantity for 60 μL of primer mix | |
| IGHV1 | CAGGTGCAGCTGGTGCAGTCTGG | 10 | |
| IGHV2 | CAGGTCAACTTAAGGGAGTCTGG | 10 | |
| IGHV3 | GAGGTGCAGCTGGTGGAGTCTGG | 10 | |
| IGHV4 | CAGGTGCAGCTGCAGGAGTCGGG | 10 | |
| IGHV5 | GAGGTGCAGCTGTTGCAGTCTGC | 10 | |
| IGHV6 | CAGGTACAGCTGCAGCAGTCAGG | 10 | |
| 5’ IGHV leader primers | |||
| IGHV1L | AAATCGATACCACCATGGACTGGACCTGGAGG | 5 | |
| IGHV1bL | AAATCGATACCACCATGGACTGGACCTGGAG(C/A) | 5 | |
| IGHV2aL | AAATCGATACCACCATGGACACACTTTGCT(A/C)AC | 5 | |
| IGHV2bL | AAATCGATACCACCATGGACATACTTTGTTCCAC | 5 | |
| IGHV3aL | AAATCGATACCACCACCATGGAGTTTGGGCTGAGC | 5 | |
| IGHV3bL | AAATCGATACCACCACCATGGA(A/G)(C/T)T(G/T)(G/T)G(G/A)CT(G/C/T)(A/C/T)GC | 5 | |
| IGHV4L | AAATCGATACCACCATGAAACACCTGTGGTTCTT | 10 | |
| IGHV5L | AAATCGATACCACCATGGGGTCAACCGCCATC | 10 | |
| IGHV6L | AAATCGATACCACCATGTCTGTCTCCTTCCTC | 10 | |
| 3’ IGHJ primers | |||
| IGHJ1-2 | TGAGGAGACGGTGACCAGGGTGCC | 20 | |
| IGHJ3 | TGAAGAGACGGTGACCATTGTCCC | 10 | |
| IGHJ4-5 | TGAGGAGACGGTGACCAGGGTTCC | 20 | |
| IGHJ6 | TGAGGAGACGGTGACCGTGGTCCC | 10 | |
Table 1. Primer sequences and quantities for the PCR amplification of the clonotypic IGHV-IGHD-IGHJ gene rearrangement.
| Reagent | Volume (μL) |
| RB x 10 buffer | 5 |
| MgCl2 (50 mM) | 3 |
| dNTPs (10 mΜ) | 2 |
| Primer IGHV leader/FR1 mix (10 μM) | 3 |
| Primer IGHJ mix (10 μM) | 3 |
| H2O | 31.5 |
| Total volume | 47.5 |
Table 2. Reagents for the PCR amplification of the clonotypic IGHV-IGHD-IGHJ gene rearrangement.
| Temperature | Duration | Cycle number | Description |
| 94 °C | 5 minutes | 1 | polymerase activation |
| 94 °C | 1 minute | 39 | denaturation |
| 59 °C | 1 minute | annealing | |
| 72 °C | 1.5 minutes | extension | |
| 72 °C | 10 minutes | 1 | final extension |
| 18 °C | ∞ | 1 | preservation |
Table 3. Thermal cycling conditions for the PCR amplification of the clonotypic IGHV-IGHD-IGHJ gene rearrangement.
| Temperature | Duration | Cycle number | Description |
| 94 °C | 5 minutes | 1 | polymerase activation |
| 96 °C | 20 seconds | 30 | denaturation |
| 50 °C | 20 seconds | annealing | |
| 60 °C | 4 minutes | extension | |
| 4 °C | ∞ | 1 | preservation |
Table 4. Thermal cycling conditions for the IG gene sequencing reaction.
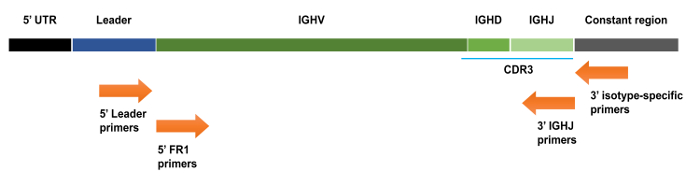
Figure 1. Schematic representation of an IGHV-IGHD-IGHJ gene rearrangement with various primer annealing locations indicated. 5' IGHV primers anneal to the leader sequence that is located upstream of the IGHV coding sequence. 5' IGHV framework (FR) primers anneal to the start of the FR1 region, which is located within the rearranged IGHV gene, whereas the 3' IGHJ primers anneal to the end of the rearranged IGHJ gene. UTR: untranslated region. Please click here to view a larger version of this figure.
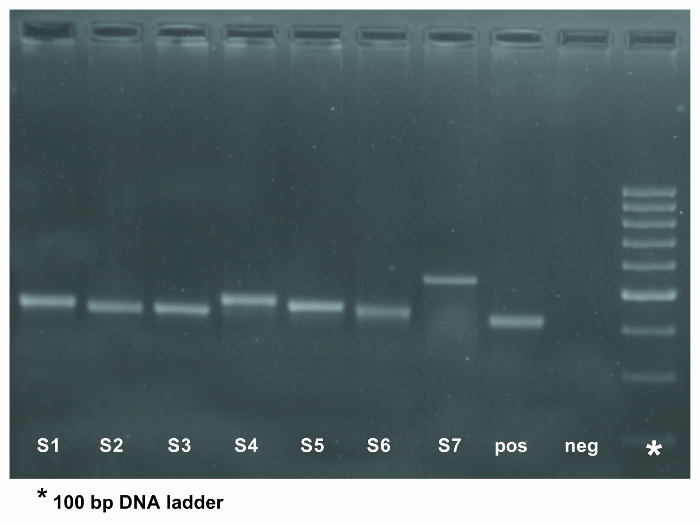
Figure 2. PCR amplification of the IGHV-IGHD-IGHJ gene rearrangement in 7 patient samples using 5' IGHV leader primers. The eighth lane represents a positive control whereas the negative control for the PCR was loaded into the ninth lane. The expected PCR product is approximately 500 base pairs in size when using IGHV leader primers. The asterisk in the last column of the gel indicates the 100 bp DNA ladder. Please click here to view a larger version of this figure.
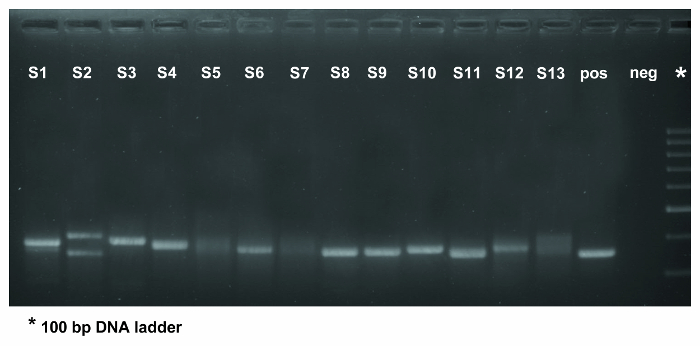
Figure 3. PCR amplification of the IGHV-IGHD-IGHJ gene rearrangement in 13 patient samples using IGHV FR1 primers. The fourteenth lane contains the positive control whilst the negative control was loaded into the fifteenth lane. As evidenced in lane 2, for which DNA was the starting material, several PCR bands are observed, which should be excised and sequenced seperately. Samples in lanes 5, 7, and 13 yielded polyclonal results (evidenced by the smear in the gel) and, thus, the PCR step should be repeated. If a second PCR fails to amplify the rearranged IG gene(s) the analysis should be performed on a new sample (if possible) or using a different primer set. The expected PCR product is approximately 350 base pairs in size when using IGHV FR1 primer mixes. The asterisk in the last column of the gel indicates the 100 bp DNA ladder. Please click here to view a larger version of this figure.
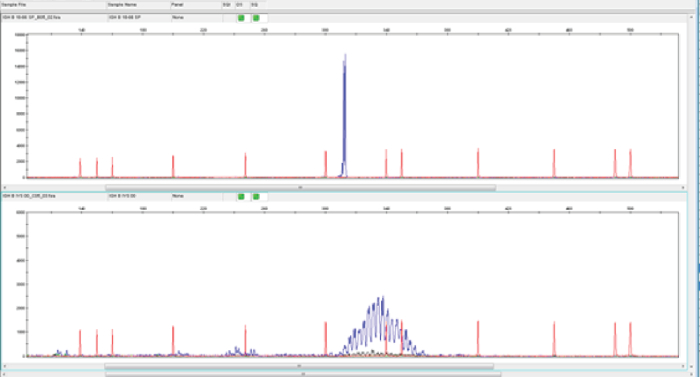
Figure 4. Assessment of clonality using genescan analysis. In genescan analysis, monoclonal PCR products give rise to a dominant peak of fluorescent products of identical size (upper panel), whereas polyclonal PCR products result in a more normal distribution of product sizes (lower panel). The red traces are peaks from a fluorescently-labeled DNA ladder that is loaded in the same capillary injection as the sample being analysed. The size standard fragments are subjected to the same electrophoretic force as the sample and are therefore exposed to the same injection conditions. The uniform spacing of the size standard fragments confirms precise size calling. The blue traces represent the monoclonal (top panel) or polyclonal (bottom panel) nature of the samples. Please click here to view a larger version of this figure.
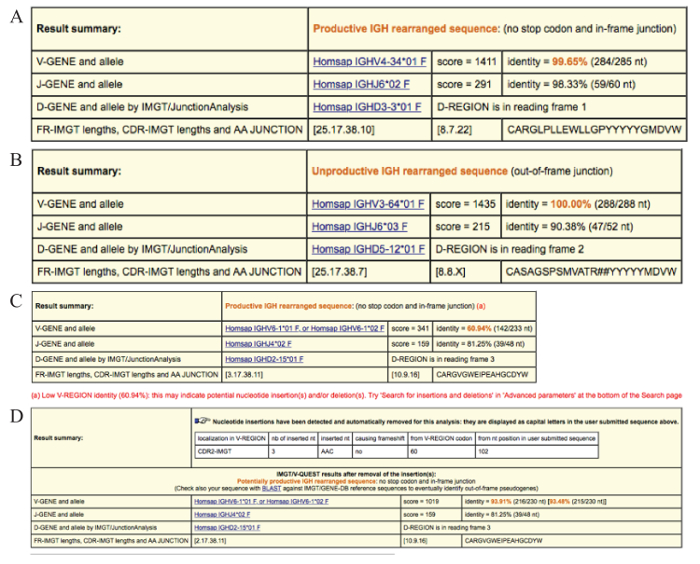
Figure 5. Examples of the result summary tables provided by IMGT/V-QUEST. (A) Result summary table for a productive IGHV-IGHD-IGHJ gene rearrangement (no stop codon(s) and an in-frame sequence junction). The V-GENE and allele and the J-GENE and allele identified in the user sequence by IMGT/V-QUEST in this case are IGHV4-34*01 and IGHJ6*02 (on the basis of the highest alignment score evaluation). The percent identity is 99.65% due to a single somatic mutation. The D-GENE and allele identified by IMGT/JunctionAnalysis is the IGHD3-3*01 in reading frame 1. The lengths of the 4 framework regions (FRs) as well as the 3 complementarity determining regions (CDRs) are indicated in brackets. The amino acid (AA) sequence of the IGHV-D-J junction is also provided. In this example the junction comprised 22 AAs. (B) Result summary table for an IGHV-IGHD-IGHJ gene rearrangement sequence that is unproductive due to an out-of-frame junction. An X for the CDR3-IMGT length indicates that for this sequence the length could not be determined. The '#' signs observed in the AA junction sequence indicate a frameshift. (C) Result summary table warning of low V-REGION identity (60.94%) and indicating that the 'insertions and deletions' option in the 'Advanced parameters' section should be used. (D) Result summary table for the case with germline low percent identity illustrated in (C) after repeating the sequence analysis using the 'Search for insertions and deletions' option. The correct identity percent is displayed in brackets and is calculated considering the insertion as a single mutational event. Please click here to view a larger version of this figure.
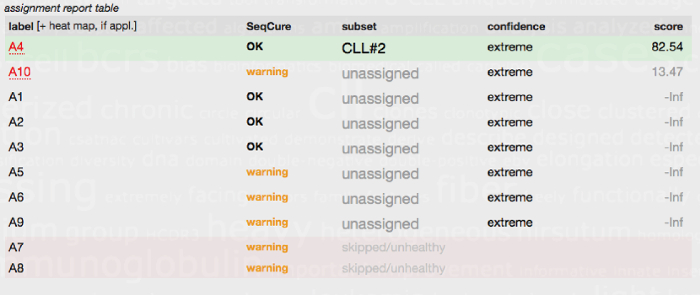
Figure 6. Subset assignment report table generated by the ARResT/AssignSubsets tool. The FASTA IG sequences from 10 CLL patients (A1-A10) were submitted to the ARResT/AssignSubsets tool. Case A4 was assigned to CLL stereotyped subset #2 (patients within this group are known to have a poor prognosis irrespective of SHM load) with high confidence. Seven of the other cases/sequences were not assigned to a major stereotyped subset and hence classed as unassigned. Two of the submitted sequences (A7 and A8) could not be used for subset assignment and were instead classed as skipped/unhealthy due to issues with the sequence, i.e., due to an out-of-frame junction or the presence of stop codons. A comprehensive explanation of the table headings and the tool is available at the ARResT/AssignSubsets website32. Please click here to view a larger version of this figure.
