Preparation of Drosophila Larval Samples for Gas Chromatography-Mass Spectrometry (GC-MS)-based Metabolomics
Summary
This protocol describes how to prepare Drosophila larvae for GC-MS-based metabolomic analysis.
Abstract
Recent advances in the field of metabolomics have established the fruit fly Drosophila melanogaster as a powerful genetic model for studying animal metabolism. By combining the vast array of Drosophila genetic tools with the ability to survey large swaths of intermediary metabolism, a metabolomics approach can reveal complex interactions between diet, genotype, life-history events, and environmental cues. In addition, metabolomics studies can discover novel enzymatic mechanisms and uncover previously unknown connections between seemingly disparate metabolic pathways. In order to facilitate more widespread use of this technology among the Drosophila community, here we provide a detailed protocol that describes how to prepare Drosophila larval samples for gas chromatography-mass spectrometry (GC-MS)-based metabolomic analysis. Our protocol includes descriptions of larval sample collection, metabolite extraction, chemical derivatization, and GC-MS analysis. Successful completion of this protocol will allow users to measure the relative abundance of small polar metabolites, including amino acids, sugars, and organic acids involved in glycolysis and the TCA cycles.
Introduction
The fruit fly Drosophila melanogaster has emerged as an ideal system for studying the molecular mechanism that regulate intermediary metabolism. Not only are most metabolic pathways conserved between Drosophila and humans, but key nutrient sensors and growth regulators, such as insulin, Tor, and myc, are also active in the fly1,2. As a result, Drosophila can be used to explore the metabolic basis of human diseases ranging from diabetes and obesity to neurodegeneration and cancer. In this regard, Drosophila larval development provides the ideal framework in which to study a metabolic program known as aerobic glycolysis, or the Warburg effect. Just as many tumors use aerobic glycolysis to generate biomass from carbohydrates, so to do Drosophila larvae activate aerobic glycolysis to promote developmental growth3,4,5. These similarities between larval and tumor metabolism establish Drosophila as a key model for understanding how aerobic glycolysis is regulated in vivo.
Despite the fact that the fly has emerged as a popular model for studying metabolism, most Drosophila studies rely on methods that are designed to measure individual metabolites3, such as trehalose, triglycerides, or ATP. Since a specific protocol is required to measure each metabolite, assay-based studies are labor-intensive, expensive, and biased towards those compounds that can be measured using commercial kits. A solution to these limitations has emerged from the field of metabolomics, which provides a more efficient and unbiased means of studying Drosophila metabolism. Unlike an assay-based study, a single metabolomic analysis can simultaneously measure hundreds of small molecule metabolites and provide a comprehensive understanding of an organism's metabolic status6,7. This technique has significantly expanded the scope of Drosophila metabolic studies and represents the future of this emerging field8.
Metabolomic studies are primarily conducted using three technologies: (i) nuclear magnetic resonance (NMR), (ii) liquid chromatography-mass spectrometry (LC-MS), and (iii) gas chromatography-mass spectrometry (GC-MS)9. Each approach offers distinct advantages and disadvantages, and all of these technologies have been used to successfully study Drosophila metabolism. Since the research conducted in our lab is focused on small, polar metabolites, we primarily employ a GC-MS-based method. GC-MS provides the user with a number of advantages, including high reproducibility, peak resolution, sensitivity, and the availability of a standard electron impact (EI) spectral library, which allows for the rapid identification of discovered metabolic features10,11. The preparation of samples for GC-MS, however, is somewhat complex and requires a careful attention to detail. Samples must be collected, washed, weighed, and frozen in a manner that quickly quenches metabolic reactions. Furthermore, the fly carcass is resistant to standard homogenization protocols and requires a bead mill to ensure optimal metabolite extraction. Finally, samples analyzed by GC-MS must undergo chemical derivatization prior to detection12. While previously published methods describe all of these steps3,13,14, a visual protocol that would allow the novice user to reproducibly generate high quality data is still needed. Here we demonstrate how to prepare Drosophila larval samples for GC-MS-based metabolomics analysis. This protocol allows the user to reproducibly measure many of the small polar metabolites that compose central carbon metabolism.
Protocol
1. Egg Collection
- Collect adult males and virgin females of the desired genotypes. Individually age these animals in a food vial with standard Bloomington media for 3–5 days.
- Set up the appropriate matings by transferring 50 virgin females and 25 males to a new food vial.
NOTE: A minimum of six independent matings should be set up for each genotype. Only one sample will be collected from each mating (i.e., six samples collected from six independent matings). - Prepare molasses egg-laying caps.
- Mix 115 mL of molasses and 29 g of agar with 700 mL of H2O in a 2 L flask.
- Boil the molasses agar mixture on a hot plate.
- Cool to 70 °C.
- Add 25 mL of acid mix (20 mL of 85% phosphoric acid and 209 mL propionic acid in 1 L of H2O; Store in a brown bottle) and 10 mL of 10% p-hydroxy-benzoic acid methyl ester in 95% ethanol.
- Pour the molasses agar into both the lid and bottom portion of a 35 x 10 mm2 tissue culture dish.
NOTE: Before pouring molasses agar plates, be sure that the lids and/or base of the 35 x 10 mm2 plate fit snuggly into the mouth of a Drosophila stock bottle (described in Step 1.6). Depending on the brand of plates used, the base might not be usable.
- Make fresh yeast paste by adding distilled water to active dry yeast (5 g yeast/7 mL water) until the mixture becomes a paste that can be easily spread on a solid surface (i.e., the consistency of peanut butter).
- Spread approximately 1.5 g of yeast paste on the surface of a molasses egg-laying cap.
- Poke four air holes into opposite sides of a plastic 6 oz. Drosophila stock bottle using a 22 G needle.
- Transfer the newly mated flies from the food vial into culture bottles.
NOTE: Flies can be transferred by either using CO2 as a temporary anesthetic or by holding the top of vial in mouth of the bottle and sharply tapping the bottom of the bottle against a benchtop. - Quickly place a molasses egg-laying cap with yeast paste on the surface into the mouth of a Drosophila stock bottle. Use laboratory tape to secure the egg-laying cap in place.
NOTE: If flies were anesthetized prior to transfer, the bottle should be placed horizontally on the benchtop until all flies have recovered. Once flies are completely mobile, place the bottle in an incubator with the molasses egg-laying cap at the bottom of the culture. - Replace the molasses egg-laying cap at least once a day for the first two days by inverting the stock bottle, tapping the bottom of the bottle against the benchtop, removing the old egg cap, and immediately placing a new egg cap into the mouth of the bottle. Discard the old egg-laying cap.
NOTE: This step ensures that females are not holding their eggs and allows for collection of synchronized populations. - Place a new egg-laying cap into the stock bottle after day 3, replace after 2 h, and discard. Remove and replace the second egg-laying cap after four hours. Label the bottom of this egg-laying cap with the date, time, and genotype.
NOTE: The eggs collected during this 4 h window will be used for analysis. This synchronization step is critical for analyzing specific developmental stages. Eggs can be collected in this manner for several days. - Insert the egg-laying caps into a 60 x 15 mm2 Petri plate and place into an incubator at the desired temperature and humidity.
- Inspect the egg-laying caps every day for problems with starvation, population density or contamination.
NOTE: Larvae must have access to adequate food to ensure continuous development. If necessary, fresh yeast paste can be spread on all egg-laying caps that will be used in the experiment. In addition, if the larval population density is too high, larvae will begin to wander out of the culture plate. These samples should not be used for metabolomic analysis because the high population density and intermediate bouts of starvation experienced while wandering will result in inconsistent data. A density of ~80–100 middle second instar larvae (L2) per plate or 40–50 middle third instar larvae (L3) per plate is recommended. Samples that are visibly contaminated with bacteria or fungi should be discarded.
2. Larval Sample Collection
- Age larvae until the desired stage. If collecting L3 larvae, resynchronize samples at the L2–L3 molt. Resynchronization is commonly achieved using a previously described method that uses the anterior spiracles as a developmental milestone15. Alternatively, mid-L3 larvae can be synchronized using a sgs3 reporter gene16.
NOTE: In this regard, we find mid-L2 larvae (~60 h after egg laying) are ideally suited for most analyses because larval development is still relatively synchronous at this stage, carefully collected samples have adequate food to develop to this timepoint, and the number of animals per sample is manageable (see below). An additional synchronization step is necessary for L3 larvae because the numerous ecdysone pulses that occur during L3 development have dramatic effects on metabolism and will generate artifacts in unsynchronized populations. - Use a dissecting needle to gently lift the yeast paste off the agar. Place the yeast onto a new molasses agar cap.
NOTE: Most larvae will eat at the interface between the molasses agar and the yeast paste. - Use a small paintbrush to collect larvae from the newly exposed surface of the molasses agar. Place the larvae into a 1.5 mL centrifuge tube on ice.
NOTE: Appropriate sample sizes for each developmental stage are as follows: 50 first instar larvae (L1); 25 mid-L2 larvae; 20 early L3 larvae; 15 mid-L3 or late L3 larvae. - Wash and freeze samples (steps 2.3 through 2.8) in small batches (four to six samples) while collecting a large sample set. Samples must be frozen within 20 min of placing larvae in tubes.
NOTE: We recommend using Eppendorf 1.5 mL Flex Tubes because other tubes may interfere with the sample transfer at step 3. Samples must be collected in microfuge tubes. Do not collect samples in the bead tubes described in step 3.1. Ceramic beads retain the NaCl wash solution, which results in inaccurate mass measurements and introduces contaminants into the sample. - Add 1 mL of ice-cold 0.9% NaCl into the tube, close the lid, and vertically flip the tube to thoroughly to wash the larvae.
- Place the tube back on ice for ~30 s. During this time, larvae will sink to the bottom of the tube, but yeast will remain in suspension. Once all larvae have formed a loose pellet, remove the NaCl solution using a 1 mL pipette.
- Repeat step 2.4 and step 2.5 twice, or until the final wash solution is clear.
Note: Additional wash steps may be necessary if the collected sample contains an excessive amount of yeast. - Centrifuge the samples at 2,000 × g for 1 min at 4 °C.
- Remove all residual solution using a 200 µL pipette.
- Immediately freeze the sample in liquid nitrogen.
NOTE: The protocol can be paused at this step. Samples can be stored for up to 3 months at -80 °C.
3. Transfer of Samples to Bead Tubes
- Each larval sample must be transferred from the 1.5 mL microfuge tube to a 2 mL screwcap tubes containing 1.4 mm ceramic beads. In preparation for this transfer step, use an ethanol-proof marker to label the side of a 2 mL screwcap bead tube (markings on the lid will be destroyed during step 4.4).
- Tare the mass of the labeled bead tube using an analytical balance capable of accurately measuring 0.01 mg.
- If larval samples were stored at -80 °C, place sample tubes in liquid nitrogen prior to transfer.
- Use long forceps to remove the 1.5 mL sample tube from the liquid nitrogen dewar.
- Wearing nitrile gloves, grab the frozen tube, invert, and sharply pound the tube lid against the benchtop to dislodge the frozen pellet. Immediately pour the pellet into a pre-tared 2 mL screwcap bead tube. If the pellet fails to dislodge, return the tube to liquid nitrogen and repeat.
NOTE: Larval pellets will not dislodge from all microfuge tubes. If using a tube other than the recommended one, determine if larval pellets can be dislodged prior to collecting samples for analysis. - Quickly measure the combined mass of the larval pellet and bead tube. Immediately place the sample tube into liquid nitrogen. The larval pellet mass will be used to normalize the metabolomic data.
NOTE: The protocol can be paused at this step. Samples can be stored for up to 3 months at -80 °C.
4. Sample Extraction
- Place the sample tubes in a -20 °C benchtop cooler.
- Add 0.8 mL of prechilled (-20 °C) 90% methanol containing 2 µg/mL succinic-d4 acid into each tube. Return the sample to the -20 °C benchtop cooler.
NOTE: The succinic-d4 acid serves as an internal standard. Only use HPLC grade H2O and methanol. Since methanol and other organic solvents are volatile and difficult to accurately measure using air displacement pipettes, we recommend using positive-displacement pipettes for this step and all following steps. - Set up a negative control by adding 0.8 mL of prechilled (-20 °C) 90% methanol containing 2 µg/mL succinic-d4 acid into an empty bead tube.
- Homogenize samples for 30 second at 6.45 m/s using a bead mill homogenizer located in a 4 °C temperature control room.
NOTE: Failure to rapidly and completely homogenize the larval pellet is a common source of variability in metabolomics analysis. Only use homogenizers that are capable of destroying frozen larval tissues within 30 s. - Return the homogenized samples tubes to the -20 °C benchtop cooler and incubate them in a -20 °C freezer for at least 1 h.
- Centrifuge the tubes at 20,000 × g or maximum speed for 5 min at 4 °C to remove the resulting precipitate.
- Transfer 600 µL of the supernatant into a new 1.5 mL microcentrifuge tube. Do not disturb the precipitate. If the precipitate pellet becomes dislodged while pipetting the supernatant, return all supernatant to the tube and repeat step 4.6.
- Place the sample tubes in a vacuum centrifuge. Be sure that all tubes are open before sealing the centrifuge. Dry the samples at room temperature until all solvent is removed (this step usually takes ~16 h to complete).
NOTE: If necessary, the protocol can be paused at this step. Dried sample can be stored at -80 °C.
5. Chemical Derivatization
- If the dried samples were stored at -80 °C, place unopened sample tubes in a vacuum centrifuge and dry for 30 min. This step must be performed prior to opening the sample tube.
NOTE: This step removes the condensation that collects on the outside of the microfuge tube when removed from the freezer. This portion of the protocol is extremely sensitive to H2O and extra precaution must be taken to keep all reagents as dry as possible. - Prepare a solution of 40 mg/mL methoxylamine hydrochloride (MOX) in anhydrous pyridine. Only use anhydrous pyridine. Store MOX and pyridine in a desiccator and prepare the MOX solution daily.
CAUTION: MOX and pyridine are toxic. Prepare this solution in the fume hood.- Use a heat gun to dry a 1 mL glass syringe.
- Insert the needle of the syringe into the bottle of anhydrous pyridine. Remove 1 mL of pyridine and add to a microfuge tube containing 40 mg of MOX.
- Flush the bottle of anhydrous pyridine with argon. Seal the bottle and return to the desiccator.
- Dissolve the MOX in the pyridine by incubating the tube in a thermal mixer at 35 °C for 10 min at 600 rpm.
- Add 40 µL of 40 mg/mL MOX in anhydrous pyridine solution to the dried sample.
- Vortex for 10 s and briefly centrifuge (10,000 x g for 20 s).
- Incubate at 30 °C for 1 h at 600 rpm in a thermal mixer.
- Centrifuge at 20,000 × g or maximum speed for 5 min to remove the particle matter.
- Transfer 25 µL of supernatant into an autosampler vial with a 250 µL deactivated glass microvolume insert.
- Add 40 µL of N-methyl-N-trimethylsilyltrifluoracetamide (MSTFA) containing 1% TMCS.
CAUTION: MSTFA is toxic. Perform this step in the fume hood.
NOTE: If available, this step can be completed by a Gerstel autosampler. - Place a cap on the autosampler vial and seal using a crimper tool.
- Incubate the sample at 37 °C for 1 hour with shaking (250 rpm).
- Prepare a fatty acid methyl ester standard (FAMES) solution as previously desceribed3. Add 3 µL of FAMES to the autosampler vial using a robotic autosampler immediately prior to injection.
NOTE: FAMEs are used to calibrate the retention time shift and check the instrument performance. If no robotic autosampler is available, FAMEs can be added during step 5.8.
6. GC-MS Detection
NOTE: In most cases, the user will conduct this step with the assistance of a mass spectroscopy core facility. This protocol is designed to be used with a 30 m, GC column with a 5 m guard column.
- Randomize the sample order.
- Prepare the GC-MS.
- Set the helium carrier gas flow rate to 1 mL/min.
- Set the inlet temperature to 250 °C.
- Program the GC to execute the following temperature gradient:
- Initial temperature to 95 °C with a hold of 1 min.
- Increase the temperature to 110 °C at the rate of 40 °C/min with a hold of 2 min.
- Increase to 250 °C by 5°C/min ramp.
- Increase to 330 °C at the rate of 25 °C/min with a final hold of 4 min.
- Set solvent delay to 3.5 min.
NOTE: The time can be changed according to the GC-MS system. The purpose of this step is to prevent MSTFA and MOX from damaging the detector.
- Inject 1 µL of the derivatized sample into the GC-MS (split ratio of 10:1).
NOTE: Inject 2 µL of sample if the intensity of peaks is too low. The injection order of samples should be randomized. - Operate the mass spectrometer in full scan mode over a mass range of 50–500 m/z.
7. Data Analysis
- Analyze metabolomic data using either a targeted or untargeted approach. Targeted analysis is focused on measuring the abundance of a defined set of metabolites, such as the lactate measurements that we describe below. In contrast, untargeted analysis uses an unbiased approach to identify any metabolic feature that is significantly changed between two sample sets.
NOTE: Our lab primarily uses the free programs MetAlign17 and MetaboAnalyst18,19 for data analysis. Since an adequate description of both the quality control, normalization, and data processing steps are beyond the scope of this manuscript, we refer the user to more detailed protocols that are devoted to data processing20,21,22. In addition, a flow chart describing the steps required for this analysis can be found elsewhere8.
Representative Results
Lactate dehydrogenase (dLDH) mutants, which lack dLDH activity4, and genetically-matched controls were collected as mid-L2 larvae and processed according to protocol described above. When compared with controls, mutant larvae exhibit significant changes in lactate, pyruvate, and L-2-hydroxyglutarate4. Spectra were acquired with an Agilent GC6890-5973i MS system. An example of the GC-MS spectra generate with our protocol is shown in Figure 1. There are many visible features and a notable peak for trehalose, which normally represents the largest peak in a larval sample and is usually oversaturated (Figure 1A,B). The individual spectrum of negative control sample is shown in Figure 1C. Although there are still several visible peaks in the negative control sample, the intensity and the number of peaks are reduced when compared with the experimental samples shown in Figure 1A,B. These peaks primarily result from column bleed, the internal standard (succinic-d4 acid), FAMEs, and contaminating fatty acids. Failed sample preparation will generate a spectrum similar to the one shown in Figure 1C.
All the spectra were preprocessed using MetAlign17 and data were normalized with the internal standard and pellet mass. Subsequently, the data were submitted to MetaboAnalyst18,19 for statistical analysis. Principle component analysis (PCA) clearly shows that the two groups separate from each other and that there is no outlier in either group (Figure 2A). Further analysis shows significant changes in the metabolites known to be affected by loss of dLDH (Figure 2B)4.
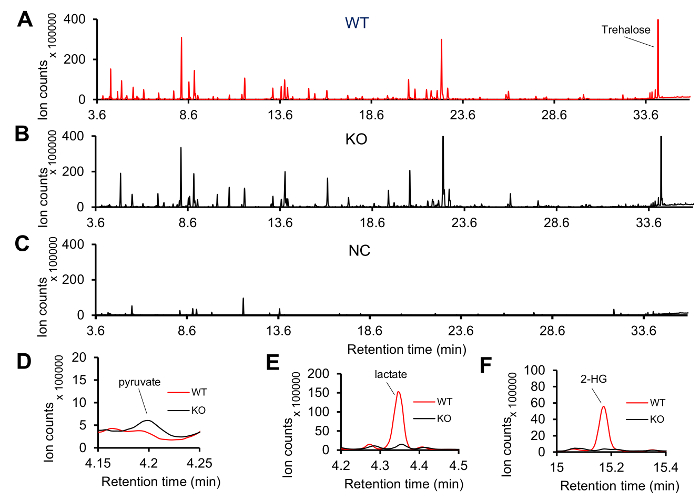
Figure 1: Representative GC-MS spectra of Drosophila larval extract. (A-B) Typical spectra of mid-L2 larval extracts from wild type (WT) and dLDH mutants (KO). (C) A representative GC-MS spectrum generated from a negative control (NC) sample. Most of the peaks in this spectrum are from internal standard, FAMEs and fatty acids. (D-F) When compared with WT samples, the KO samples exhibit elevated levels of (D) pyruvate and decreased levels of (E) lactate and (F) 2-hydroxyglutarate (2-HG). Please click here to view a larger version of this figure.
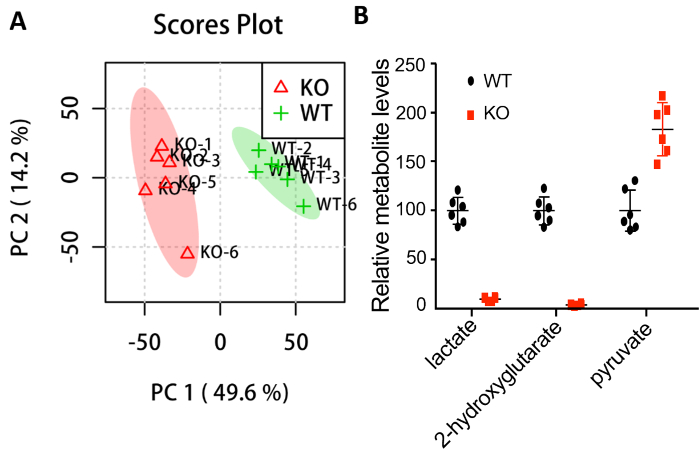
Figure 2: Statistical analysis shows the different metabolic profiles between wild type (WT) and dLDH knockout (KO) larvae. (A) PCA scores plot. (B) Metabolites that exhibit significant changes in dLDH mutants. All data points are plotted relative to the mean of the WT control, which was adjusted to an arbitrary value of 100. Prior to analysis, data was normalized to the succinic-d4 acid internal standard and larval pellet mass. Data shown as mean ± one standard deviation. p <0.01 for all metabolites using a two-tailed t-test. Please click here to view a larger version of this figure.
Discussion
Metabolomics provides an unparalleled opportunity to survey the metabolic reactions that compose intermediary metabolism. The sensitivity of this technology, however, renders data susceptible to genetic background, developmental cues, and a variety of environmental stresses, including temperature, humidity, population density, and nutrient availability. Therefore, a high quality and reproducible metabolomics analysis requires that samples be collected under highly controlled conditions. While several reviews emphasize this point3,8,23, here we provide a step-by-step method for collecting larvae that is designed to ensure reproducibility.
The most common source of variability in a metabolomics analysis stems from a failure to quench, or stop, metabolic reactions after the sample is collected. In this regard, metabolism will stop upon freezing in liquid nitrogen and metabolic enzymes will be destroyed when the sample is homogenized in -20 °C methanol. Assuming that the user pays special attention to keeping a sample frozen prior to methanol extraction, our protocol is designed to ensure that the metabolomics data generated by this procedure represent an accurate snapshot of larval metabolism. Should the user experience unacceptable levels of data variability, the user should reexamine the collection and metabolite extraction protocol with special attention paid to ensuring (1) that metabolism is rapidly quenched (Steps 2.9–4.2) and (2) that the sample is efficiently homogenized in 90% methanol (Step 4.4). In this regard, many bead mills provide insufficient force to homogenize samples within the specified time and we encourage the user to use an instrument that has been used in previous studies8.
Our protocol also highlights key steps that the novice user must note in order to reproducibly derivatize samples. This is essential for GC-MS because many metabolites have limited volatility or poor thermal stability. Derivatization increases both the volatility and thermal stability of many metabolites, thereby increasing the number of compounds that can be reproducibly measured. As a result, samples that have undergone incomplete derivatization will exhibit non-reproducible peak areas, heights, and shapes. As we emphasize here, a common source of failed derivatization is exposure of the sample to water, which decreases the chemical derivatization efficiency. Therefore, all reagents, including MSTFA, MOX, and pyridine, must be kept under moisture-free conditions. The need to maintain dry conditions also extends to preparing samples that are stored at -80 °C, which should be placed in a vacuum centrifuge for 30 min before opening to ensure that condensation does not enter the sample. We would also like to emphasize that GC-MS is a sensitive technology, and as a result, larger samples will not necessarily generate better data. Our protocol provides experimentally tested sample sizes and will reproducibly measure most metabolites in central carbon metabolism. Some of these metabolites are very abundant and increased sample size will lead to signal saturation in the spectrum. In fact, the signal for trehalose is already saturated in our analysis and a split ratio of 100:1 for GC-MS injection must be used to accurately measure this compound. Finally, the user should note that because we use a polar extraction solution, our protocol is not appropriate for lipid extraction. Our protocol also does not account for fatty acids contaminants that could be present on the plastic tubes and tips, which is why some fatty acid signals appear in the negative control sample (Figure 1B).
Finally, the methods detailed here provides a powerful tool for studying Drosophila metabolism. This protocol, however, is not specific to Drosophila and can be used with few modifications for conducting metabolic studies in any small invertebrate. Regardless of the species, this simple method allows for relative quantification of nearly all amino acids, intermediates in glycolysis and the TCA cycle, as well as a number of other small polar molecules. When combined with the unparalleled genetic tools available to the Drosophila community, this type of metabolomic analysis places the fly at the forefront of metabolic research for the foreseeable future.
Declarações
The authors have nothing to disclose.
Acknowledgements
Thanks to members of the Indiana University Mass Spectroscopy Facility and the University of Utah Metabolomics Core Facility for assistance in optimizing this protocol. J.M.T. is supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119557.
Materials
| Unsulfured blackstrap molasses | Good Food, INC | ||
| Drosophila Agar Type II | Genesee Scientific | 66-103 | |
| Pyridine | EMD Millipore | PX2012-7 | |
| Methoxyamine hydrocholoride (MOX) | MP Biomedicals, LLC | 155405 | |
| MSTFA with 1% trimethylchlorosilane | Sigma | 69478 | |
| Fleischmann’s Active dry yeast | AB Mauri Food Inc | 2192 | |
| 6oz Drosophila stock bottle | Genesee Scientific | 32-130 | |
| Soft tissue homogenizing mix (2 mL tubes) | Omni International | SKU:19-627 | |
| Vial insert, 250 µL deactivated glass with polymer feet | Agilent | 5181-8872 | |
| Succinic acid-2,2,3,3-d4 | Sigma | 293075 | |
| SpeedVac | Thermo | SC210A | |
| o-Phosphoric acid | Fisher Scientific | A242-1 | |
| Propionic acid | Sigma | P5561 | |
| p-Hydroxy benzoic acid methyl ester | Genesee Scientific | 20-258 | |
| Bead Ruptor | Omni International | SKU:19-040E | |
| ThermoMixer F1.5 | Eppendorf | 5384000012 | |
| MultiTherm Shaker with a 24 X 12 mm block | Benchmark Scientific | H5000 | |
| Methanol | Sigma | 34860 | |
| 1.5 mL centrifuge tube | Eppendorf | 22364111 | |
| Falcon 35 X 10 mm tissue culture dish | Corning Incorporated | 353001 | |
| GC column | Phenomex | ZB-5MSi |
Referências
- Owusu-Ansah, E., Perrimon, N. Modeling metabolic homeostasis and nutrient sensing in Drosophila: implications for aging and metabolic diseases. Disease Models & Mechanisms. 7 (3), 343-350 (2014).
- Sieber, M. H., Spradling, A. C. The role of metabolic states in development and disease. Current Opinion in Genetics & Development. 45, 58-68 (2017).
- Tennessen, J. M., Barry, W. E., Cox, J., Thummel, C. S. Methods for studying metabolism in Drosophila. Methods. 68 (1), 105-115 (2014).
- Li, H., et al. Drosophila larvae synthesize the putative oncometabolite L-2-hydroxyglutarate during normal developmental growth. Proceedings of the National Academy of Sciences of the United States of America. 114 (6), 1353-1358 (2017).
- Tennessen, J. M., Baker, K. D., Lam, G., Evans, J., Thummel, C. S. The Drosophila Estrogen-Related Receptor Directs a Metabolic Switch that Supports Developmental Growth. Cell Metabolism. 13 (2), 139-148 (2011).
- Nicholson, J. K., Lindon, J. C., Holmes, E. Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 29 (11), 1181-1189 (1999).
- Fiehn, O. Metabolomics – the link between genotypes and phenotypes. Plant Molecular Biology. 48 (1-2), 155-171 (2002).
- Cox, J. E., Thummel, C. S., Tennessen, J. M. Metabolomic Studies in Drosophila. Genética. 206 (3), 1169-1185 (2017).
- Lenz, E. M., Wilson, I. D. Analytical strategies in metabonomics. Journal of Proteome Research. 6 (2), 443-458 (2007).
- Pasikanti, K. K., Ho, P. C., Chan, E. C. Y. Gas chromatography/mass spectrometry in metabolic profiling of biological fluids. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 871 (2), 202-211 (2008).
- Want, E. J., Nordstrom, A., Morita, H., Siuzdak, G. From exogenous to endogenous: The inevitable imprint of mass spectrometry in metabolomics. Journal of Proteome Research. 6 (2), 459-468 (2007).
- Garcia, A., Barbas, C., Metz, T. O. . Metabolic Profiling: Methods and Protocols Vol. 708 Methods in Molecular Biology. , 191-204 (2011).
- Chan, E. C. Y., Pasikanti, K. K., Nicholson, J. K. Global urinary metabolic profiling procedures using gas chromatography-mass spectrometry. Nature Protocols. 6 (10), 1483-1499 (2011).
- Dunn, W. B., et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature Protocols. 6 (7), 1060-1083 (2011).
- Ashburner, M. . Drosophila: A Laboratory Manual. , 171-178 (1989).
- Biyasheva, A., Do, T. V., Lu, Y., Vaskova, M., Andres, A. J. Glue secretion in the Drosophila salivary gland: a model for steroid-regulated exocytosis. Biologia do Desenvolvimento. 231 (1), 234-251 (2001).
- Lommen, A. MetAlign: Interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Analytical Chemistry. 81 (8), 3079-3086 (2009).
- Xia, J., Wishart, D. S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Current Protocols in Bioinformatics. 55, (2016).
- Xia, J., Sinelnikov, I. V., Han, B., Wishart, D. S. MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Research. 43 (W1), W251-W257 (2015).
- Lommen, A. Data (pre-)processing of nominal and accurate mass LC-MS or GC-MS data using MetAlign. Methods in Molecular Biology. 860, 229-253 (2012).
- Xia, J., Wishart, D. S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Current Protocols in Bioinformatics. 55, (2016).
- Xia, J., Wishart, D. S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nature Protocols. 6 (6), 743-760 (2011).
- Li, H., Tennessen, J. M. Methods for studying the metabolic basis of Drosophila development. Wiley Interdisciplinary Reviews Developmental Biology. 6 (5), (2017).
